




Dermatite atópica é manifestação cutaneomucosa crônica, recorrente e de caráter multifatorial resultante da interação entre elementos ligados, principalmente, ao déficit da barreira cutânea, da homeostase da resposta imune, de aspectos neurológicos e de padrões de reatividade a antígenos ambientais, que se estabelece em indivíduos predispostos geneticamente. Além da pele, a diátese atópica compromete outros órgãos como as vias aéreas (superiores e inferiores), olhos, trato digestório e envolve aspectos neuropsiquiátricos, que infligem morbidade adicional ao paciente dermatológico. Os diferentes fenótipos da doença dependem, fundamentalmente, da participação de cada um desses fatores, em diferentes circunstâncias da vida, como faixas etárias, padrões de exposição ocupacional, atividade física, poluição, carga genética e fatores climáticos. O melhor entendimento da complexidade de sua patogênese torna possível não somente a compreensão dos alvos terapêuticos, mas como identificar elementos preponderantes que medeiam a atividade da doença em cada circunstância, para melhor indicação das estratégias de tratamento e mitigação de fatores desencadeantes. Esta revisão narrativa apresenta uma atualização na patogênese da dermatite atópica, especialmente direcionada para o entendimento das manifestações clínicas, dos principais fenótipos da doença no contexto das estratégias terapêuticas disponíveis.
Dermatite atópica (DA), ou eczema atópico, é doença inflamatória crônica, recorrente e multifatorial caracterizada por eczema, pele seca (xerose) e prurido em graus variados, mas em geral, intenso, que compromete diferentes dimensões da qualidade de vida: sintomático, funcional, emocional, relações interpessoais e profissional. Em virtude de sua alta prevalência, especialmente na infância, além da qualidade de vida, inflige impacto na dinâmica familiar e custos ao sistema de saúde.1–4
O conhecimento científico acerca dos diferentes aspectos da DA e da diátese atópica evoluiu consideravelmente nos últimos anos. Sua patogênese é complexa, envolvendo mecanismos imunomediados, e seu entendimento vem avançando em relação a predisposição genética, alterações estruturais e funcionais da barreira epidérmica, respostas imunes inatas e adaptativas, colonização da pele por microrganismos bactérias e fungos, reatividade aos ácaros da poeira domiciliar, elementos neurocomportamentais e gatilhos para exacerbação da doença subclínica.5
Numerosos fatores de base genética e estímulos ambientais interagem, no mesmo indivíduo, levando ao desencadeamento ou agravamento da DA. Tal complexidade de elementos justifica a variabilidade de sua expressão fenotípica, intensidade e curso clínico. Fatores étnicos, geográficos, climáticos, poluentes e ocupacionais compõem as principais circunstâncias que contribuem para tal variabilidade na apresentação clínica da DA. Há ainda variação no quadro clínico relacionado à faixa etária do doente, o que também guarda correlação com o padrão de exposição ambiental e outas condições atópicas (asma e rinite alérgicas, alergia alimentar, conjuntivite alérgica e esofagite eosinofílica).5,6
Este artigo constitui revisão narrativa multidisciplinar sobre aspectos atuais da patogênese da DA, que, apesar de não totalmente elucidada, pode ser compreendida a partir da interação de aspectos singulares como: predisposição genética; alterações da barreira cutânea; ativação dos queratinócitos por estímulos estressores mecânicos, físicos ou químicos; desequilíbrio de respostas da imunidade inata e adaptativa/perda da tolerância imune; fatores intrínsecos e extrínsecos como desencadeantes ou agravantes da doença; comorbidades orgânicas associadas e sensibilização das vias neurais do prurido, de modo que se estabelece uma interação bidirecional neural e imune na DA.5,7
Diátese atópicaHipersensibilidade é a comorbidade mais comum da DA. Cerca de 70% das crianças com DA estão sensibilizadas a um ou mais antígenos. Além disso, a diátese atópica apresenta forte carga genética. A maioria das crianças com DA tem pelo menos um dos genitores com alergia respiratória, cutânea ou ambos. O filho de um dos genitores alérgicos tem duas a três vezes mais chances de desenvolver atopia, o que aumenta para três a cinco vezes mais quando ambos os genitores são alérgicos.8
A principal via para sensibilização e perpetuação das lesões de hipersensibilidade induzida por alimentos é cutânea. A via gastrintestinal, principalmente no intervalo entre o 4° e o 6° mês de vida, é geralmente associada à indução de tolerância, já que células T e B regulatórias foram descritas desempenhando um papel na regulação da patogênese da DA e alergia alimentar, bem como no desenvolvimento de tolerância aos alimentos envolvidos.9,10
A disfunção da barreira cutânea que ocorre na DA aumenta a permeabilidade a alérgenos e irritantes, contribuindo para a perda da hidratação e persistência de inflamação cutânea subclínica.9,11 A formação de lesão inflamatória subclínica e a absorção de antígenos alimentares por via cutânea, seja por contaminação na poeira ou por uso de produtos que contenham proteínas alimentares, leva à sensibilização nos bebês com DA.9
Com base nos achados de alteração da barreira e desenvolvimento de alergia alimentar, idealizou‐se o uso profilático de emolientes em crianças com DA e risco de alergia alimentar; porém, o estudo BEEP mostrou que o emoliente pode aumentar o risco de DA em virtude da colonização de patógenos na pele, e a colonização por Staphylococcus aureus está associada a maior risco de alergia alimentar.11
Em modelo murino, a pele colonizada por S. aureus leva a ativação de células apresentadoras de antígeno dérmicas após a exposição da pele inflamada a alérgenos alimentares exógenos, expansão de células B IgE+ do centro germinal e geração de IgE específica para alérgenos alimentares, além de citocinas inflamatórias (IL‐4, IL‐5, linfopoetina estromal tímica [TSLP] e IL‐33). Além disso, demonstrou‐se que a exposição epicutânea combinada à enterotoxina B estafilocócica aumentou as respostas alergênicas ao ovo e amendoim. Assim, a disbiose microbiana cutânea também contribui com a disfunção da barreira, modulando a sensibilização alérgica.12
Portanto, lactentes com eczema são expostos ao antígeno alimentar via pele, induzindo células imunes e produzindo anticorpos IgE (de sensibilização alérgica).13 Isso implica a importância não só de induzir tolerância imunológica oral, mas também prevenir a sensibilização alérgica através da pele, a fim de reduzir as demais hipersensibilidades.10 Na Inglaterra, foi ainda demonstrado que a alergia ao amendoim estava associada ao uso de produtos para a pele contendo óleo de amendoim.10 É importante ressaltar que, no Brasil, muitos óleos utilizados na hidratação cutânea contêm antígenos alimentares como amendoim, coco, amêndoas, uva, entre outros.
No estudo HealthNut, 5.276 crianças com DA foram monitoradas durante os primeiros quatro anos de vida, demonstrando aumento do risco de desenvolver alergias alimentares, como a alergia ao amendoim e ovo.13 Uma revisão sistemática envolvendo 66 estudos revelou que a sensibilização alimentar em crianças com DA era seis vezes maior em comparação com controles saudáveis aos 3 meses de idade.10
Diversos estudos confirmaram associação entre alergia alimentar e quadro mais grave de DA, bem como ligação entre o desenvolvimento precoce e persistente de DA e alergias alimentares, observando maior probabilidade de sensibilização a alimentos comuns, como leite, ovo e amendoim. Em suma, a DA de início precoce é relacionada ao desenvolvimento de alergia alimentar. Por outro lado, a alergia alimentar agrava a DA ou aumenta o risco de comorbidades, incluindo anafilaxia.8,10,14
Alergia das vias aéreas superiores e inferiores são habituais nos pacientes com DA. Dermatophagoides pteronyssinus, D. farinae, pólens e baratas são os alérgenos mais relevantes. Entretanto, como a DA apresenta diferentes perfis de resposta imune, são frequentes os casos sem associação com asma e rinite, especialmente nas DA de início tardio, com resposta mais celular do que dependente da IgE.15
A exposição aos aeroalérgenos pode desencadear reações cutâneas na DA, sugerindo contato direto com a pele como a principal via de estímulo. Existem vários mecanismos possíveis para os aeroalérgenos desencadearem a DA. Ao penetrar na barreira cutânea comprometida da DA, os alérgenos intactos podem ser capturados, processados e apresentados por células apresentadoras de antígenos via apresentação facilitada por IgE.14 O complexo IgE‐antígeno é então internalizado e degradado, com subsequente apresentação de antígeno no complexo principal de histocompatibilidade classe II para os linfócitos T.14 De fato, os valores de IgE sérica se associam com a atividade da doença respiratória, mas não cutânea.14 Entretanto, os alérgenos podem também ser fagocitados e apresentados para reconhecimento pelos linfócitos T, respectivamente, sem envolvimento de IgE.15
A DA também está associada a doenças da superfície ocular (DSO). Entre 25% e 50% dos doentes com DA grave (Odds Ratio/razão de chances [OR=2,17]), asma e rinite (OR=1,76) e DA de início na infância (OR=1,34) são associados à DSO. O mecanismo imunopatogênico envolve imunidade adaptativa de células T contra alérgenos ou patógenos na superfície ocular. Clinicamente, DSO inclui conjuntivite, ceratoconjuntivite seca, blefarite, ceratocone e ceratite infecciosa e não infecciosa (inflamação corneana); a conjuntivite é a mais comum entre todas.16
A conjuntivite apresenta subtipos. A mais comum é a alérgica (sazonal e perene), seguida por papilar, vernal (inflamação crônica da borda externa da mucosa conjuntival palpebral), blefaroconjuntivite atópica (pela infamação da pálpebra e conjuntiva circunjacente), ceratoconjuntivite atópica e infecciosa. A ceratoconjuntivite atópica está associada a risco de dano corneano progressivo e perda visual. Há elevado risco de conjuntivite bacteriana (pelos Staphylococcus spp.) e vírus (adenovírus e Herpes simplex). Conjuntivites no doente com DA requerem abordagem multidisciplinar pelo risco de comprometimento visual.16
O tratamento com biológicos anti‐IL‐4/IL‐13 associa‐se à inflamação conjuntival, especialmente após três meses de uso do dupilumabe, em uma restrita porção de pacientes adultos (6%–30%), o que ocorre especialmente quando são observados elevados níveis séricos basais da Thymus and Activation‐Regulated Chemokine/CCL17 (TARC) e IgE total. Esses casos evoluem com diminuição das células globosas epiteliais na conjuntiva, com redução na produção da mucina, aumento da infiltração de células imunes, instabilidade do filme lacrimal, disfunção de barreira e inflamação conjuntival.16
A DA costuma ser o primeiro passo no desenvolvimento da denominada “marcha atópica”, já que a disfunção da barreira epidérmica favorece a permeação de antígenos associados ao seu início. A marcha atópica refere‐se à progressão sequencial para rinite alérgica, alergia alimentar ou asma; entretanto, mesmo tal progressão tem base genética. Em estudo conduzido nos EUA, com 18.596 crianças com DA, observou‐se que as crianças afro‐americanas tinham progressão a partir da DA para asma, enquanto as de ascendência euro‐americana progrediam de DA para rinite alérgica, e as crianças asiáticas tinham marcha de DA para alergia alimentar IgE‐mediada. O risco de asma entre crianças de ascendência afro‐americanas foi seis vezes maior do que entre as crianças com ascendência euro‐americana.17
A alarmina TSLP produzida pelos queratinócitos submetidos a estresse, alérgenos ou patógenos ocasiona a ativação de células dendríticas e secreção de quimiocinas, que atraem linfócitos T auxiliadores tipo 2 (Th2) à pele, liberando citocinas pró‐alergênicas (citocinas de inflamação tipo 2, como IL‐4, IL‐13, Il‐5, IL‐31). Na DA, a produção desregulada e excessiva de citocinas, em especial a IL‐4 e IL‐13, ocasiona a inibição da expressão de proteínas epidérmicas como a filagrina, loricrina e involucrina, o que reforça os defeitos na barreira epidérmica, que favorecem a permeação de alérgenos e patógenos, retroalimentando a doença. A resposta desregulada de citocinas de inflamação tipo 2 ocorre em diferentes sítios anatômicos do corpo. Ao nível dos brônquios, IL‐4 e IL‐13 atuam mediando a inflamação e alterações de remodelamento das vias aéreas, predispondo o desenvolvimento da asma tipo 2‐relacionada (50%–70% dos asmáticos).18
No contexto da rinossinusite crônica (RSC), o dano da barreira epitelial das vias aéreas superiores inicia e perpetua a inflamação do tipo 2, e fatores ambientais como vírus e outros estímulos geram alarminas como TSLP, IL‐25 e IL‐33, que aumentam a inflamação nasal. Entre pacientes asmáticos, há prevalência em torno de 40% de RSC com polipose – a DA é mais comum entre esses doentes com RSC com polipose nasal, em relação àqueles com RSC sem polipose. Acredita‐se que esse mecanismo de inflamação tipo 2 seja compartilhado simultaneamente, predispondo e amplificando a resposta imune do hospedeiro a estímulos externos, como ocorre na conjuntivite alérgica e na esofagite eosinofílica.18
Genética da dermatite atópicaA DA é mais prevalente em famílias com a doença; fatores genéticos respondem por 82% do risco de desenvolvimento de DA, versus 18% dos fatores ambientais.19 Ademais, crianças com ambos genitores com histórico de atopia apresentam chance cinco vezes maior de desenvolver DA com fenótipo de início precoce e persistente, comparado com crianças com genitores sem histórico.20
A herdabilidade de DA em gêmeos monozigóticos e dizigóticos é 72%–86% e 21%–23%, respectivamente.21 O subgrupo clínico mais associado à transmissão genética é o de pacientes com início precoce e resolução na adolescência, além do sexo masculino.22
A alta variação de prevalência e as diferentes características da diátese atópica associadas às etnias, como o aumento da heritabilidade genética de asma em crianças negras e baixa prevalência de DA em populações centro‐asiáticas, estimularam dezenas de estudos em DA e grupos populacionais.23,24 Apesar disso, são necessárias pesquisas de maior amplitude genômica para direcionar, rigorosamente, como alterações genéticas de base ancestral atuam em uma interface genético‐social.25
Um estudo com randomização mendeliana identificou associação entre idade de início de diabetes e também uso de álcool com DA.26 Já uma revisão sistemática recente de 30 estudos com randomização mendeliana determinou que índice de massa corporal, flora intestinal, refluxo gastroesofágico e a via da IL‐18 eram fatores causais de DA, enquanto DA era fator causal de comorbidades como insuficiência cardíaca, artrite reumatoide e conjuntivite.27
No Brasil, a prevalência geral de DA em comunidades indígenas (1,9%) mostrou‐se inferior à de população de cidades populosas, apesar de não ser estratificado por idade e de haver a expectativa de maior proporção de crianças nessas áreas menos urbanizadas.2,28
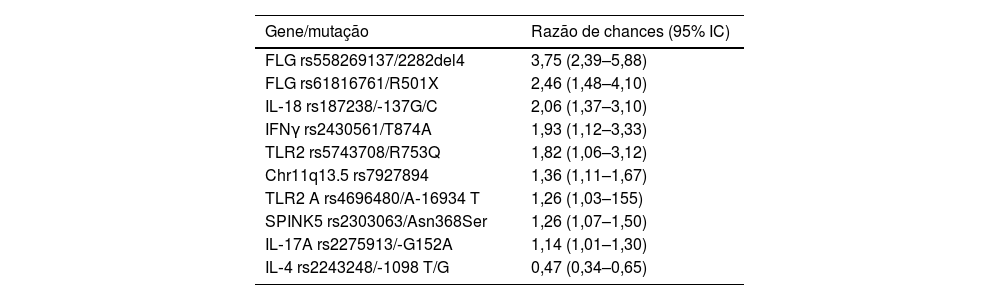
Associação de genes candidatosHá dezenas de loci de susceptibilidade à DA caracterizados em diversas populações, que medeiam funções como resposta a interferona gama (IFNγ), imunidade inata, funções dos linfócitos T e disfunções da barreira epidérmica (tabela 1).26,29,30 Do mesmo modo, as mais relevantes associações de vias de sinalização, analisadas pela base de dados Reactome, foram as de IL4/IL‐13, IL‐2 e IL‐12.31 Estima‐se que até 15% da heritabilidade da DA seja determinada por variantes comuns e 12,5% por variantes raras nesses loci.32
Principais mutações associadas à dermatite atópica, independentemente da população estudada30
| Gene/mutação | Razão de chances (95% IC) |
|---|---|
| FLG rs558269137/2282del4 | 3,75 (2,39–5,88) |
| FLG rs61816761/R501X | 2,46 (1,48–4,10) |
| IL‐18 rs187238/‐137G/C | 2,06 (1,37–3,10) |
| IFNγ rs2430561/T874A | 1,93 (1,12–3,33) |
| TLR2 rs5743708/R753Q | 1,82 (1,06–3,12) |
| Chr11q13.5 rs7927894 | 1,36 (1,11–1,67) |
| TLR2 A rs4696480/A‐16934 T | 1,26 (1,03–155) |
| SPINK5 rs2303063/Asn368Ser | 1,26 (1,07–1,50) |
| IL‐17A rs2275913/‐G152A | 1,14 (1,01–1,30) |
| IL‐4 rs2243248/‐1098 T/G | 0,47 (0,34–0,65) |
O gene da filagrina (FLG) apresenta grande diversidade de mutações genéticas etno‐dependentes e é o gene mais estudado em estudos de DA; certas variações são associadas a formas mais graves da doença.31 Em populações altamente miscigenadas, como a brasileira, podem ser identificadas mutações associadas a diferentes ancestralidades.33
Além disso, o FLG apresenta mutações associadas a início mais precoce da doença (assim como o NLRP10), enquanto outros SNP de genes como AFF1 e EHMT1 aumentam a suscetibilidade para início mais tardio da DA.34,35 Uma metanálise de estudos de gene candidatos em europeus e asiáticos confirmou a associação entre variantes de FLG e o risco de DA; além disso, revelou novos loci associados em europeus, IL‐18 e TGFB1, e em asiáticos, IL12RB1 e MIF.36
Alterações no gene SPINK5 também cursam com modificações na barreira cutânea, uma vez que está relacionado ao inibidor endógeno de protease LEKTI, que exerce importante papel na homeostase da barreira da pele, incluindo descamação da epiderme e constituição da barreira lipídica. Diversos polimorfismos de SPINK5 estão associados à incidência da DA em chineses.37
Quanto a fatores ambientais, identificou‐se associação entre DA e poluição do ar em 19 genes ligados ao estresse oxidativo, que estão associados principalmente à ativação de neutrófilos e regulação de linfócitos T helper e linfócitos T reguladores (TREGs).38
Estudos de genoma completo também identificaram genes associados a fenótipos de complicações da DA como o eczema herpético, como SIDT2 e RBBP8NL, que participam na resposta dos queratinócitos à infecção do herpes vírus tipo 1.39 Além disso, o eczema paradoxal em pacientes com psoríase em uso de imunobiológicos foi associado a variantes que podem influenciar a expressão de transcritos ou a sequência de aminoácidos na via IL‐12, mas não nas vias IL4/13 e IL17/23. Do mesmo modo, os autores especulam que a inibição da IL‐12 por variantes genéticas dessa via habilita o desenvolvimento da resposta Th2, o que inicia o eczema paradoxal.40
Fatores epigenéticosEpigenética é o conjunto de mudanças na expressão gênica que podem ser herdados ou não e que, entretanto, não alteram a sequência do DNA. Estímulos ambientais (p. ex., poluição, dieta, tabagismo, radiação, agentes químicos) são os principais reguladores epigenéticos. Os principais estudos epigenéticos em DA são relacionados à metilação de DNA de dois genes, NLRP2 (envolvido com atividade de macrófagos) e PITPNM2 (relacionado à neutrófilos), ambos associados ao tabagismo. Ainda a associação de eczema herpético e níveis séricos de IgE com a metilação nos genes IL‐4 e IL‐13.41–43 Além disso, há microRNAs que estão sobre ou infrarregulados na pele ou soro de pacientes com DA, como o MiR‐144, associado ao início precoce da doença.31
Barreira cutâneaA pele constitui o maior órgão do ser humano e desempenha a função de barreira de proteção, separando o corpo do ambiente externo, o que resguarda o organismo de agressões térmicas, actínicas, osmóticas, químicas, infecciosas e mecânicas. Epiderme, derme e tecido subcutâneo em conjunto com glândulas écrinas, folículo pilossebáceo e outros apêndices cutâneos formam uma estrutura integrada que interage promovendo essa função protetora.
Uma das principais características da barreira cutânea é sua estrutura multicamada, que desempenha funções interdependentes. O estrato córneo é a porção mais externa da pele que atua como a principal barreira física, em arranjo semelhante a tijolos (corneócitos, com característica química polar) e cimento (substância intercelular, predominantemente lipídica e de ceramidas, com características apolares), que, em conjunto com as zonas de oclusão (TJ), compostas por claudinas e ocludinas expressas também na camada granulosa da epiderme e no espaço intercelular dos queratinócitos dos canais das glândulas sudoríparas, regulam o influxo e o efluxo de fluidos e eletrólitos para dentro e para fora da pele.44
Os contatos células‐células, especialmente na epiderme humana, são críticos para a estruturação tecidual e sua homeostasia. As junções de aderência compostas por caderinas (dímeros trans e cis‐caderinas) e nectinas medeiam a adesão célula‐células. As TJ, incluindo suas claudinas, ocludinas e moléculas de adesão juncional (p. ex., ZO‐1), estabelecem a polaridade apical‐basolateral dos queratinócitos e regulam o transporte para‐celular (intercelular) de íons e solutos. A expressão de proteínas subunitárias, como claudinas e ocludinas, é reduzida pela exposição a alérgenos e cepas de estafilococos, comprometendo a integridade da barreira na DA.45,46
Assim, a estrutura epidérmica com adesão intercelular exerce função complexa de proteção, que, junto com o sistema imune cutâneo, contribui para a primeira linha de defesa contra patógenos microbianos e insultos ambientais.
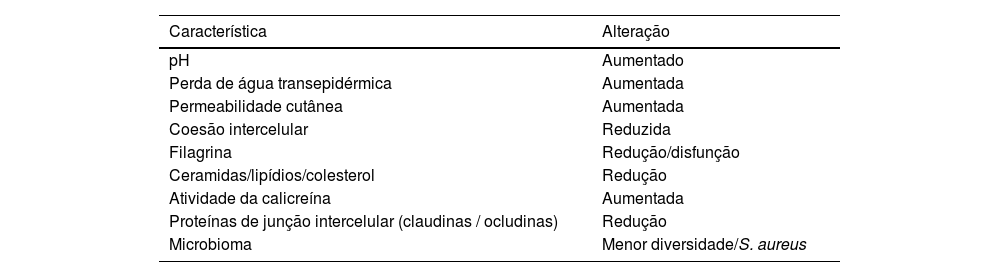
A barreira cutânea disfuncional é mais prevalente em portadores de DA (tabela 2), e seu déficit tem várias consequências clínicas, já que esses pacientes se caracterizam por alta reatividade a estímulos ambientais e patógenos, reforçando o papel do restauro da barreira cutânea e da proteção contra gatilhos no controle da doença. A perda de água transepidérmica resulta em ressecamento e descamação da pele, o pH elevado aumenta a proliferação bacteriana, enquanto a menor adesão entre os corneócitos favorece a penetração de alérgenos e irritantes, podendo desencadear resposta inflamatória exacerbada.47
Características da barreira cutânea na dermatite atópica
| Característica | Alteração |
|---|---|
| pH | Aumentado |
| Perda de água transepidérmica | Aumentada |
| Permeabilidade cutânea | Aumentada |
| Coesão intercelular | Reduzida |
| Filagrina | Redução/disfunção |
| Ceramidas/lipídios/colesterol | Redução |
| Atividade da calicreína | Aumentada |
| Proteínas de junção intercelular (claudinas / ocludinas) | Redução |
| Microbioma | Menor diversidade/S. aureus |
As funções de barreira em pacientes com DA variam em diferentes fases da doença. Utilizando medições do pH e perda de água transepidérmica (TEWL), observaram‐se diferenças desses parâmetros na pele entre áreas eczematosas e aparentemente saudáveis nos pacientes com DA. Notavelmente, mesmo a pele não afetada nos pacientes com DA apresentou diferenças em comparação com os indivíduos saudáveis. Na DA, há aumento médio de pH de 4,99 para 5,68 e a TEWL passa de 6 para 30g/m2h.48
Um dos principais fatores genéticos associados à DA é a mutação no gene FLG, cujo prejuízo de função leva ao comprometimento da barreira cutânea. A filagrina é uma subunidade monomérica de uma proteína embalada em grânulos querato‐hialinos, produzidos nos queratinócitos terminais, envolvida na manutenção da barreira de permeabilidade cutânea.49 Essa proteína estrutural se liga aos filamentos dos queratinócitos para aumentar a densidade de feixes de filamentos, achatando os queratinócitos até sua forma terminal, crucial para a força e integridade da pele.49 A profilagrina, produzida a partir do gene FLG, é quebrada em filagrina monomérica, que por sua vez é degradada por proteases em aminoácidos livres, funcionando como fatores de hidratação natural para reter água, manter o pH da pele e fortalecer o estrato córneo, oferecendo proteção química e microbiana.49
Mutações no gene FLG podem resultar em perda funcional ou número menor de unidades de repetição intraexônicas, que levam à deficiência da proteína, alterando a morfologia dos corneócitos. Isso compromete a barreira para‐celular, permitindo a passagem de alérgenos e haptenos. Embora as mutações do FLG possam contribuir para a DA, não são suficientes para causar a doença isoladamente, pois uma parcela dos indivíduos com alelos nulos não a desenvolve.50
Cerca de 25% a 50% dos pacientes com DA apresentam uma mutação no gene FLG como fator predisponente, especialmente nos fenótipos extrínsecos.50 No entanto, sabe‐se que essa variação na perda de função do gene tem variações étnicas – na Europa, os valores chegam a 50% e na Ásia 25%; no entanto, pacientes afro‐americanos têm menor frequência de mutação, e a mutação do gene FLG2 é mais impactante para a doença.51 A metilação do FLG também está correlacionada com o risco de DA, corroborando nas contribuições epigenéticas para o desenvolvimento da doença.52
O desequilíbrio predominante de Th2 na DA é implicado na redução da expressão de FLG, contribuindo para o defeito da barreira. Além disso, a expressão de proteínas semelhantes, como a filagrina‐2 e a hornerina, apresenta evidências de aumentar o risco de DA.53
Outras moléculas participam do processo de formação do estrato córneo. Com a perda do núcleo, as células se achatam e as moléculas de queratina ficam paralelas, criando o envelope cornificado conectado com os lipídeos extracelulares. A força de coesão da camada córnea depende da formação de ligações covalentes de lisina e glutamina, em que proteínas precursoras são incorporadas à queratina, como involucrina, cornifina, loricrina, queratolinina e proteínas desmossômicas, como a envoplaquina e a periplaquina.54
A matriz lipídica do estrato córneo é defeituosa na pele com DA em virtude de uma alteração na expressão de enzimas envolvidas na síntese e no processamento de ácidos graxos livres (AGL) e ceramidas. Os grânulos lamelares ricos em lipídios na camada granular da epiderme fornecem o selo hidrofóbico nas camadas epidérmicas mais superficiais. A matriz lipídica estruída após o processamento por enzimas de processamento de lipídios torna‐se uma mistura de ceramidas, colesterol e AGL. Em casos de DA, há uma quantidade diminuída de ceramidas, e as presentes são mais curtas.55 Essa redução ocorre tanto na pele afetada quanto na não afetada, especialmente naqueles com anormalidades da filagrina. Além disso, foi identificada redução na relação entre ceramida e colesterol no estrato córneo nesses pacientes.56 Isso pode ser controlado pelo aumento da atividade das enzimas de processamento de lipídios ou ser resultado da inflamação Th2 subjacente, substanciando a importância de se manter a inflamação subclínica sob controle para prevenir recidivas, como demonstrado com a terapia proativa.57,58
O pH elevado do estrato córneo e a maior atividade da serina proteinase promovem a inativação e degradação da esfingomielinase ácida e da β‐glucocerebrosidase, que são as enzimas necessárias para a síntese de ceramida.59 Uma atividade elevada de serina proteinase reduz a secreção de corpos lamelares através do sinalização de ativador de plasminogênio tipo 2 e resulta na transferência anormal de várias substâncias que são secretadas do corpo lamelar.60 Isso está relacionado, em última análise, à redução de lipídeos extracelulares relatada em pacientes com DA.
Na pele lesionada, os comprimentos das cadeias de ceramida, AGL e ácidos graxos esterificados também são encurtados, o que causa anormalidades na organização lipídica epidérmica e consequente alteração na permeabilidade da barreira cutânea. Em pacientes com DA crônica, o aumento na atividade da calicreína também pode levar a essas mudanças na estrutura lipídica, induzindo a degradação da proteína de elongação de ácidos graxos de cadeia muito longa.59
Além das alterações genéticas, a inflamação crônica associada à DA favorece a disfunção da barreira cutânea, como discutido adiante. A liberação de citocinas, como IL‐4, IL‐13 e IL‐31, contribui para a alteração da diferenciação e proliferação celular na epiderme. Da mesma maneira, o tônus Th2da doença suprime a produção de peptídeos antimicrobianos e de IFN‐α, favorecendo infecções bacterianas e virais.
A disfunção da barreira cutânea, fundamentalmente, favorece o impacto de elementos ambientais na patogênese da DA. Chama‐se expossoma a interação bidirecional do ambiente externo com a saúde humana e a doença, complementando as interações ao longo da sua vida entre genes e ambiente na patogênese das doenças. Pode ser classificado como: (i) fatores externos gerais, tais como clima, biodiversidade, urbanização (fungos domiciliares e ácaros) e contextos socioeconômicos; (ii) fatores externos específicos: alérgenos, poluentes (moléculas particuladas – PM10, nanopartículas, dióxido de nitrogênio, ozônio), detergentes, metais, pólens, fumaça do tabaco, dieta, suor, fatores de estilo de vida e micróbios; e (iii) fatores internos: inflamação, metabolismo e expossoma do estresse oxidativo.61
Expossomas são os principais reguladores na interação entre a barreira epidérmica disfuncional, microbioma, genoma e desregulação imune na DA. Essa desregulação da barreira epidérmica inicia um ciclo vicioso de inflamação que se torna crônica na DA, a qual é suportada pela alteração do microbioma (disbiose) e seu desbalanço, demonstrando a importância da homeostasia da barreira epidérmica para se evitar ou restaurar a saúde cutânea na DA.61
Um exemplo de pressão ambiental que pode determinar secreção de alarminas pelos queratinócitos na epiderme é a exposição a altas concentrações de material particulado com tamanho médio <2,5μm emitidas por incêndios florestais, que rompe a barreira epidérmica por danificar proteínas estruturais (filagrina e E‐caderina) e lípides na epiderme. Além do material particulado, nanopartículas aciculares (diâmetro até 270nm) de dióxido de titânio, mas não globulares do mesmo diâmetro, foram internalizadas por queratinócitos humanos normais e produziram citocinas pró‐inflamatórias como IL1‐α, IL‐1β, IL‐6, TNF‐α e IL‐8, induzindo a inflamação cutânea.62,63
A exposição a detergentes sintéticos está associada a risco elevado de eczema relacionado a atividades laborais – em geral, profissionais da limpeza, potencialmente por meio da TEWL. Observou‐se aumento no número de casos de exacerbação da DA pelo aumento do uso de desinfetantes durante a pandemia da COVID‐19.61,64
A fumaça do tabaco, quer por exposição ativa ou passiva, entrega benzeno à pele, o que interfere na habilidade inibidora das células TREGs em secretar IL‐10 e exercer efeitos anti‐inflamatórios, o que repercute emmaior prevalência da DA em crianças com parentes tabagistas.61
Além disso, alérgenos com propriedades proteolíticas, como certos pólens e o grupo 1 dos ácaros da poeira domiciliar, promovem ruptura da barreira epidérmica e contribuem para um ciclo vicioso de inflamação‐disbiose microbiana e fragilidade da barreira cutânea.61,65 Esses elementos subsidiam terapias de dessensibilização a fim de reduzir a reatividade a esses agentes, especialmente frente a alergias respiratórias.
De modo geral, o déficit da barreira cutânea na DA é componente importante da sensibilização alérgica nesses pacientes, e também fator relevante das exacerbações da doença. Em casos com doença controlada (i.e., com inflamação subclínica), a exposição a irritantes, poeira, detergentes e banhos quentes no inverno são elementos reconhecidos como desencadeantes de recidivas clínicas, o que reforça a importância da manutenção das terapias voltadas para a recuperação da barreira cutânea e as estratégias proativas para prolongar a remissão clínica da DA.57,58
ImunopatologiaAlterações histológicasApesar de não haver achados patognomônicos, ao exame histopatológico da DA, que a discrimine das demais dermatites eczematosas, há uma série de alterações que induzem ao seu diagnóstico, em cada forma da doença, dentro de um contexto clínico.
Os achados histopatológicos variam dependendo do estágio da doença, presença de infecção, tratamento e da gravidade das lesões cutâneas. No entanto, algumas características histológicas comuns são observadas em muitos casos.
Espongiose com acantose e hiperceratose são as alterações mais características das placas eczematosas. Formas agudas, exsudativas podem conter, inclusive, vesículas intraepidérmicas, enquanto formas crônicas ou prurigoides apresentam mínima espongiose, em contraste com marcada acantose e hiperparaceratose.
O infiltrado inflamatório dérmico também varia com a forma do eczema: mais intenso na forma aguda e esparso nas formas crônicas e prurigoides. Na derme superior, predominam linfócitos típicos (especialmente CD4), especialmente perivasculares, com esparsos eosinófilos. Exocitose de linfócitos na epiderme, sem a formação de microabscessos, é achado frequente das biopsias em DA.
Vasodilatação e edema da derme superior é mais comum em lesões eczematosas agudas.
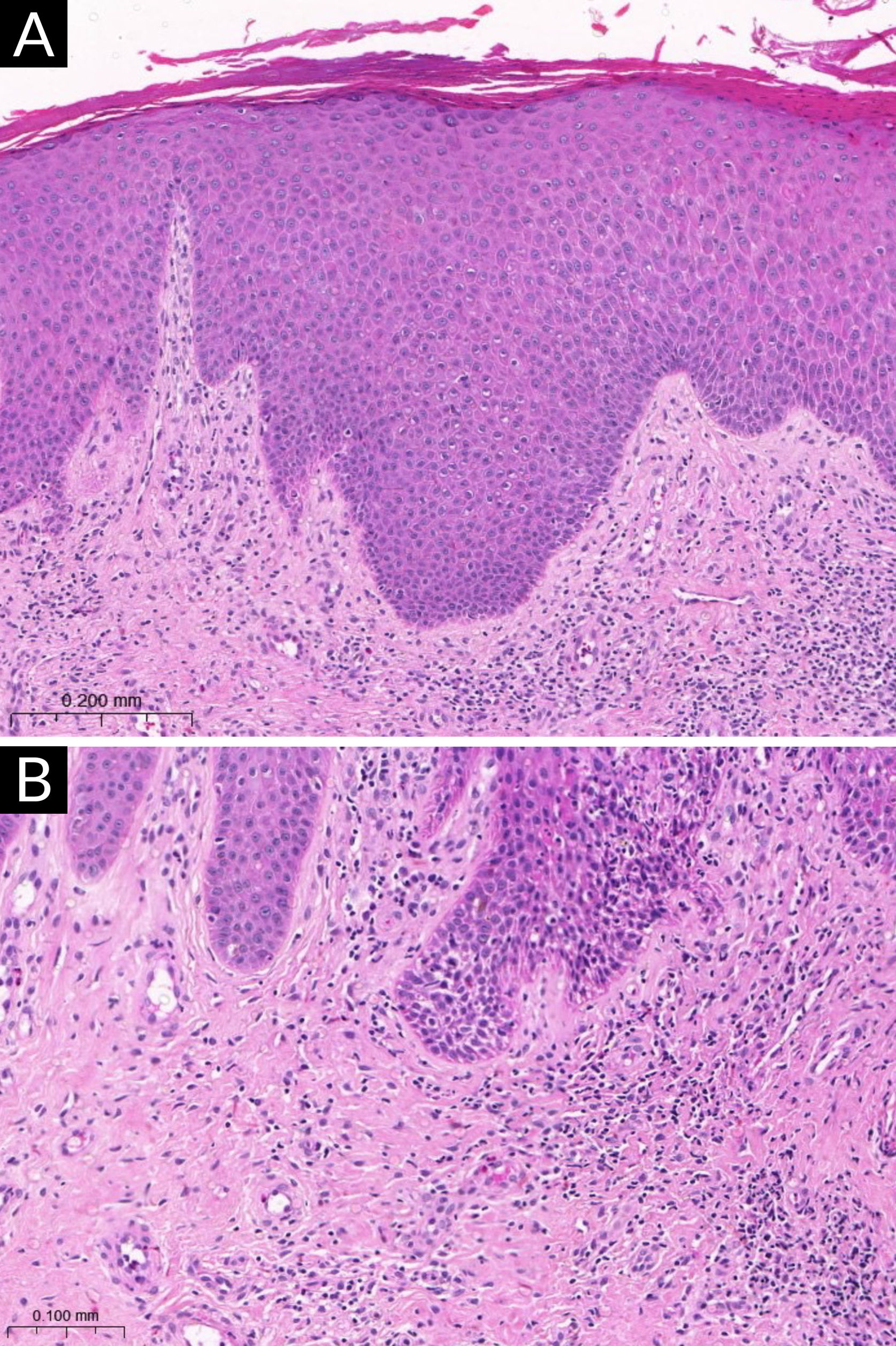
A figura 1 exemplifica uma lesão eczematosa subaguda de DA.
. (A) Epiderme com acantose irregular, hiperceratose e áreas com paraceratose, associada à espongiose discreta (Hematoxilina & eosina, 200×). (B) Infiltrado inflamatório linfocitário da derme superficial, predominantemente perivascular, com esparsos eosinófilos (Hematoxilina & eosina, 400×).")
Exame histopatológico da dermatite atópica (lesão subaguda). (A) Epiderme com acantose irregular, hiperceratose e áreas com paraceratose, associada à espongiose discreta (Hematoxilina & eosina, 200×). (B) Infiltrado inflamatório linfocitário da derme superficial, predominantemente perivascular, com esparsos eosinófilos (Hematoxilina & eosina, 400×).
A pele é um órgão imune secundário, e assim como o trato digestório e o sistema respiratório, representa uma barreira primária de reatividade contra as injúrias ambientais e patógenos, o que justifica sua alta reatividade a estímulos imunogênicos. De maneira inespecífica, acredita‐se que 95% das injúrias sejam mitigadas pela barreira cutânea, o que reforça a interação de elementos do sistema imune cutâneo que contribuem para a patogênese da DA.
Células de Langerhans (CL) são as principais apresentadoras de antígenos da epiderme, imunócitos da primeira linha de defesa, que fagocitam peptídeos a partir dos dendritos estendidos entre os queratinócitos. Também na epiderme, as células T de memória residentes propiciam resposta imune duradoura contra antígenos conhecidos, situando‐se nos locais mais prováveis de injúria ambiental e patógenos.
A derme é o microambiente imune cutâneo mais ativo. Na derme papilar, o tecido conjuntivo frouxo contém vasos e terminações nervosas, que se ramificam entre os queratinócitos. Na derme inflamada, uma diversidade de tipos celulares imunes pode ser observada, como vários subtipos de células T, macrófagos, células dendríticas, células linfoides inatas, e mastócitos, em geral localizados ao redor dos vasos sanguíneos e folículos pilossebáceos.
Como a barreira epidérmica é exposta constantemente a injúrias ambientais, as funções da epiderme como um sensor de alarme e integradora de estímulos ambientais com o sistema imune é crucial. As citocinas derivadas da epiderme, denominadas alarminas, intermedeiam uma complexa comunicação intercelular entre os queratinócitos epidérmicos e células imunes no sentido de regular a vigilância imune.
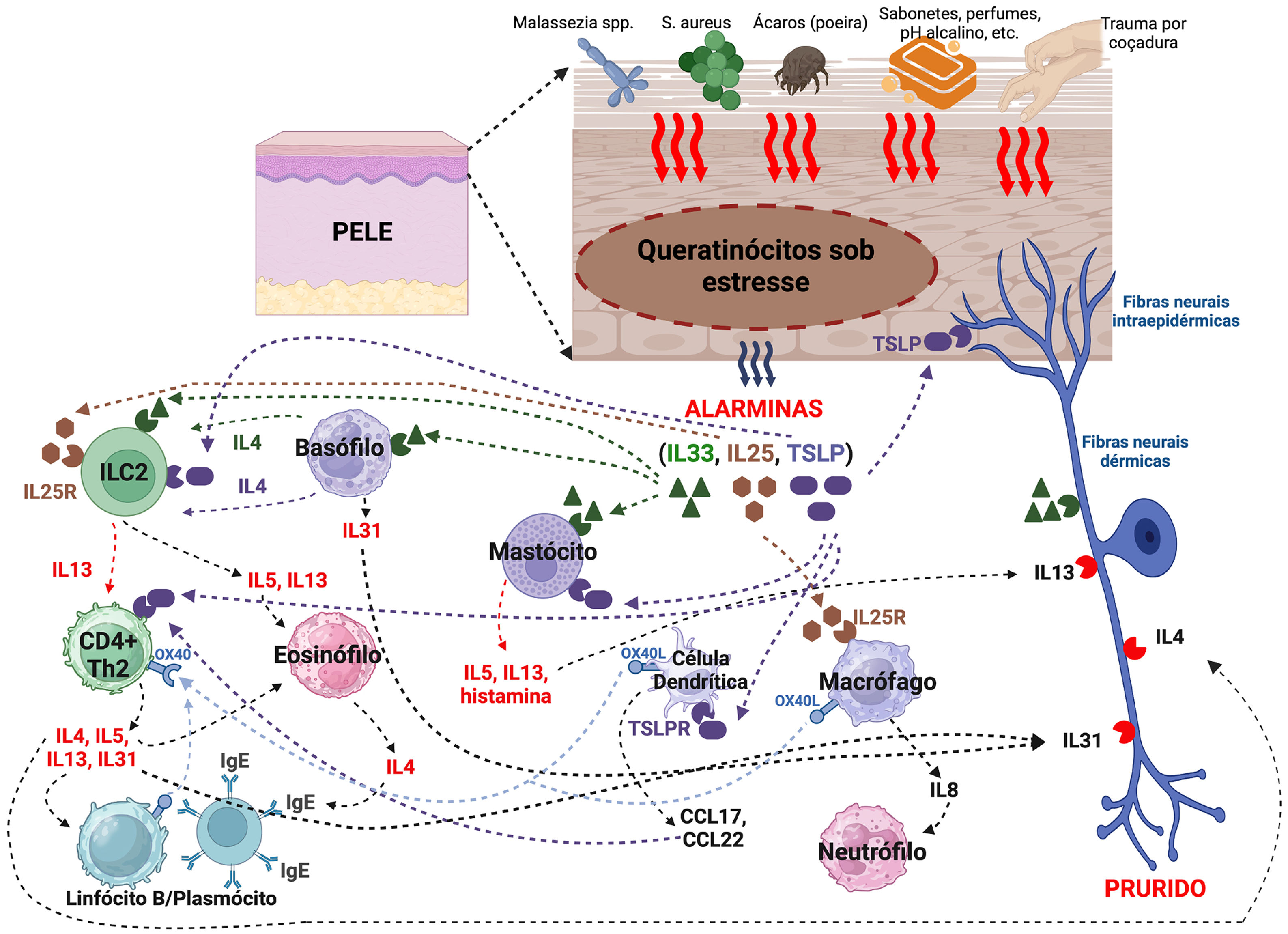
AlarminasAlarminas são moléculas endógenas que fazem parte das DAMPS (padrões moleculares associados a danos), que são importantes iniciadores da resposta inflamatória. Elas funcionam como sinais de dano (conhecidas também como estressinas) e que são liberadas para o ambiente extracelular em resposta ao dano epidérmico a fim de desencadear respostas imunes. Entre elas, há as alarminas de função tipo 2, como a linfopoietina do estroma tímico (TSLP), IL‐25 e IL‐33, as quais são orquestradoras centrais na imunidade envolvendo linfócitos T auxiliares tipo 2 (Th2). Sua desregulação ou síntese exagerada e recorrente está associada a inflamação crônica, como na DA (fig. 2).66

Uma variedade de fatores endógenos e ambientais, irritantes, poluentes e fumaça do tabaco estimulam os queratinócitos a liberar a TSLP. A ativação dos receptores de protease ativada tipo 2 (PAR2), receptores Toll‐like 4, e um membro da família dos canais receptores de potencial transitório valinoide (TRPV), incluindo o TRPV1, na membrana celular dos queratinócitos ativa a produção da TSLP por meio de fatores de transcrição, fator nuclear de células T ativadas (NFAT), fator nuclear kappa B, e fator 3 regulador da interferona.66
Variadas células hematopoiéticas e neurônios sensoriais expressam o receptor da TSLP (TSLPR). Ainda, a TSLP induz a expressão do principal complexo de histocompatibilidade de classe I e II e nas moléculas coestimuladoras nas células dendríticas, as quais produzem quimiocinas atrativas de células Th2 como a TARC e CCL22, e induzem a diferenciação Th2 por meio da expressão acentuada da molécula coestimuladora OX40L na célula dendrítica.7
A OX40 e OX40L são membros da superfamília do receptor do fator de necrose tumoral/fator de necrose tumoral (TNFR/TNF). Os membros da superfamília TNFR/TNF são moléculas de pontos de restrição imune que podem aumentar a resposta das células T por meio dessa coestimulação. A interação OX40‐OX40L torna possível uma via coestimuladora relevante na indução da expansão clonal de células CD4+ e CD8+ efetoras, intensificando a expressão de citocinas pró‐inflamatórias, e facilita a geração de células T de memória. A interação OX40‐OX40L suprime a produção de células TREG, atenuando sua função supressora na inflamação/resposta imune. Durante a fase aguda da inflamação na DA, a sinalização OX40/OX40L promove diferenciação de células Th2, que expressam mais OX40 e secretam citocinas que irão promover ruptura da barreira epidérmica, a qual ativa células apresentadoras de antígenos que subsequentemente expressam OX40L. Durante a fase crônica da DA, outros subtipos de células T expressam OX40 (Th1, Th17 e Th22), que são recrutadas para o microambiente inflamatório. Células T de memória efetoras expressam OX40 e se encontram hiperexpressas nas lesões de DA, o que pode oferecer uma explicação imune potencial para a recorrência da DA (exacerbações ou flares) e a cronicidade da DA.67
A TSLP nos queratinócitos pode ser estimulada por patógenos como S. aureus e leveduras do gênero Malassezia, além de estímulos ambientais, como radiação ultravioleta, injúria mecânica e poluentes aéreos, além da vitamina D3 pode induzir a expressão da TSLP nos queratinócitos e desenvolver um fenótipo dermatite atópica‐símile.66
A IL‐33 é predominantemente produzida pelos queratinócitos, os quais também expressam na sua membrana celular o seu receptor ST2. Ainda é produzida por fibroblastos dérmicos e macrófagos, e expressa em quantidades elevadas na DA, na qual seus níveis séricos estão elevados em comparação a controles sadios e se correlacionam com xerose e escoriações.66
A IL‐33 é liberada por queratinócitos expostos a patógenos como S. aureus e alérgenos dos ácaros da poeira domiciliar, além de estímulos ambientais como a UVB e estresse hipo‐osmótico e dano epidérmico mecânico, além da indução de INFγ e TNFα, nos queratinócitos. A IL‐33 nuclear está elevada nos queratinócitos estimulados pela TSLP, e a IL‐33 é necessária para a supressão da integridade da barreira epidérmica pela TSLP, indicando que a IL‐33 nuclear é o mediador‐chave na disfunção da barreira epidérmica induzida pela TSLP.66
A IL‐25 derivada dos queratinócitos é altamente expressa na DA, e a produção da IL‐4, IL‐13, IL‐22, endotelina‐1 e periostina aumentam a secreção da IL‐25 pelos queratinócitos. Além disso, IL‐25 atua sinergicamente com outras citocinas Th2 (IL‐4 e IL‐13) na supressão da expressão da filagrina nos queratinócitos, exacerbando os defeitos da barreira epidérmica na DA.
Alarminas ainda desencadeiam a ativação de inflamassomas na pele. Inflamassomas são complexos multiproteicos que se formam no citoplasma em resposta a sinais de perigo, e fazem parte da imunidade inata na inflamação, com a função de remover patógenos e reparar tecidos danificados. NLRP3 é o principal inflamassoma estudado na DA, cuja formação está relacionada à toxina estafilocócica e aos alérgenos dos ácaros da poeira domiciliar. As principais citocinas induzidas diretamente pela ativação de NLRP3 são IL‐1β, IL‐18, IL‐5, e IL‐31, que são associadas à resposta Th2.68 Por sua vez, a disfunção dos inflamassomas pode afetar a integridade da barreira cutânea, facilitando a penetração de alérgenos e microrganismos. Além disso, pela ação no sistema nervoso, NLRP3 está também relacionado ao comportamento de ansiedade e depressão, frequentemente encontrados na DA.69
ImunopatogêneseA patogênese da DA compreende a interação entre alterações da função de barreira, estímulos ambientais no contexto de uma resposta imune desregulada e espectral Th2 e Th17/22.61
Após ultrapassar a barreira cutânea, os antígenos são fagocitados por diferentes células apresentadoras de antígenos, especialmente as CL. Durante a inflamação cutânea, a expressão do TNFα induz a expressão da CXCL12 pelos fibroblastos dérmicos, e uma vez que as CL estejam na derme, elas produzem quantidades excessivas e expressam CCR7, entram no sistema linfático e migram aos linfonodos regionais.70
Essa contribuição das CL também ocorre no contexto da DA e alergia alimentar por meio da sensibilização transcutânea a alérgenos alimentares. Com a captura desses alérgenos pelas CL, apresentação do neoantígeno aos LT CD4+ naïve nos linfonodos de drenagem e diferenciação dos LTCD4+ em células Th2 alérgeno‐específicas secretando citocinas pró‐alergênicas (IL‐4, IL‐5, IL‐9, IL‐13), junto com as células linfoides da imunidade inata tipo 2 (ILC2), agem modulando a atividade de outras células dendríticas, células Th2, eosinófilos e mastócitos.71
Após a fase inicial do desequilíbrio da resposta imune na DA, em que alarminas, inflamassomas, células apresentadoras de antígenos epidérmicas e dérmicas desencadeiam respostas inflamatórias tipo 2 (repostas inatas com alarminas, ILC2s, e adaptativas com citocinas IL‐4, IL‐5, IL‐13, Il‐31, além da produção de IgEs alérgenos específicas), nos casos crônicos da DA, uma dominância de respostas Th1 e Th17 é gradativamente observada, mediada pelo eixo INFγ/TNFα e IL‐17, respectivamente, enquanto respostas Th22 direcionadas pela IL‐22 também estão presentes.61
Desse modo, IL‐4 e IL‐13 constituem as principais citocinas associadas à patogênese da DA inicial, pois além de promover as respostas inflamatórias tipo 2 e recrutar eosinófilos à pele, elas danificam a barreira epidérmica por suprimir a expressão de proteínas estruturais como a filagrina, loricrina e lípides, enquanto concomitantemente aumentam a deposição de colágeno na derme, o que resulta em remodelamento cutâneo e liquenificação.61
Além dessas ações, IL‐4 e IL‐13 contribuem para: (i) o prurido neurogênico pela estimulação direta nos receptores dessas citocinas nas terminações sensoriais pruritogênicas na epiderme e derme; (ii) o estímulo da produção da 3β‐hidroxiesteroide desidrogenase‐1 e andrógenos, o que resulta na diminuição da concentração dos triglicerídeos nos sebócitos e queratinócitos dentro da epiderme, contribuindo para o dano na pele atópica; e (iii) a secreção de mais IL‐13 pelos mastócitos dérmicos em resposta ao dano cutâneo e a inibição da produção de IL‐12 associada aos Th1 e liberação subsequente do INFγ, suprimindo as respostas Th1 mediadas a antígenos encontrados na DA.61
Além do dano epidérmico na DA e a exposição a alérgenos ativando respostas imunes Th2, as TREG apresentam‐se desreguladas, perpetuando o ambiente de resposta inflamatória. Tal redução e comprometimento funcional das TREG pode ser atribuída a certos componentes do expossoma, como a fumaça do tabaco na gestação e disbiose do microbioma cutâneo. Na DA, as células TREG após estimulação com superantígenos estafilocócicos (enterotoxina tipo B) perdem atividade imunossupressora.5
Elevados níveis séricos da IL‐21 são observados em pacientes com DA, a qual é primariamente produzida por células Th17, o que se associa com maior gravidade da DA, quando comparado a controles sadios, de modo que a IL‐21 promove diretamente inflamação tipo 2 e suprime respostas Th1, suprimindo a produção da INFγ.61
A IL‐22, uma citocina do polo Th22, encontra‐se elevada tanto na DA aguda quanto na forma crônica, danificando a barreira epidérmica, pela supressão de proteínas relevantes na diferenciação normal da epiderme, aumentando a sensibilização a antígenos e promovendo resposta imune exacerbada do tipo 2, além de contribuir na patogênese do prurido, pela promoção de citocinas indutoras do prurido.72
O aumento da atividade enzimática da fosfodiesterase 4 observada na DA determina redução do monofosfato de adenosina cíclica intracelular, o qual é um regulador negativo da produção de citocinas, sendo mais um elemento que contribui para a secreção aumentada de mediadores pró‐inflamatórios envolvidos na inflamação aguda e crônica da DA.61
O receptor de hidrocarboneto arílico (AhR) é um fator de transcrição que atua na regulação de múltiplas vias de sinalização envolvendo a homeostase cutânea, respostas imunes e a função da barreira epidérmica. Na pele, o AhR é expresso primariamente nas CL e nos queratinócitos. Na ausência de seus ligantes ativadores, tais como certos hidrocarbonetos aromáticos policíclicos, o AhR está associado a um complexo de proteínas plasmáticas, que incluem a proteína de choque térmico 90 e outras proteínas chaperonas. O AhR controla respostas a xenobióticos (drogas) e estressores ambientais, preservando a homeostase. Esse receptor é ativado por uma variedade de ligantes de baixo peso molecular que podem chegar até a epiderme por via endógena, pela dieta, pelo meio ambiente e por fontes bacterianas, tais como metabólitos do ácido araquidônico, compostos indigoides, metabólitos heme, fatores dietéticos como flavonoides, carotenoides e metabólitos produzidos por bactérias intestinais comensais. Do meio ambiente chegam PAH, dioxinas e bifenilados policlorinados. A ativação dos AhR por diferentes ligantes pode conduzir ao estímulo ou supressão de diferentes genes, ocasionando ampla variedade de respostas biológicas.73
A ativação fisiológica do AhR promove a diferenciação dos queratinócitos e a formação da barreira epidérmica, com a síntese de filagina, loricrina e involucrina. A exposição a poluentes ambientais, como hidrocarbonetos presentes no tabaco e material particulado sólido ou líquido que permanece na atmosfera como resultado da combustão de derivados fósseis (carvão e petróleo), ativa a via do AhR aumentando o risco de desenvolvimento da DA. A ativação do AhR na pele humana pode determinar produção excessiva da IL‐22, o que pode potencializar a atividade da DA. Na DA, o material particulado proveniente de contaminação ambiental estimula o AhR a produzir nos macrófagos e queratinócitos, de maneira deletéria, a IL‐33 (alarmina), a qual tende a induzir respostas Th2, que junto com a IL‐4 e IL‐13 suprimem a expressão da filagrina e loricrina, gerando disfunção da barreira epidérmica, e tornando‐se fonte de IL‐33 para indução do prurido ligando‐se aos receptores ST2 nas terminações nervosas sensoriais epidérmicas e dérmicas. A IL‐33, por sua vez, tem sua produção estimulada nos queratinócitos, via MAPK, por meio da retroalimentação positiva por citocinas produzidas pelas células inflamatórias dérmicas na DA, como Th1 (via produção de IIFN‐γ), Th17 (via IL‐17) e Th2 (via IL‐4 e IL‐13). Além disso, a IL‐33 reduz a síntese de IL‐37 na camada de células granulosas da epiderme, que fisiologicamente estimula a produção do complexo de diferenciação epidérmica, incluindo a filagrina e a loricrina, comprometendo ainda mais a barreira epidérmica. Coaltar, tapiranof, bem como certos flavonoides, reduzem o estresse oxidativo, suprimindo citocinas pró‐inflamatórias e estimulando a restituição da expressão de proteínas da barreira epidérmica.73
A imunidade inata e adaptativa e seus elementos celulares, tais como basófilos, eosinófilos e macrófagos, contribuem de modo relevante na patogênese da DA. Os basófilos participam do início da DA por meio do aumento da expressão da IL‐4 e suas interações com queratinócitos e macrófagos dérmicos, resultando em hiperplasia epidérmica e disfunção da barreira epidérmica. Além disso, comprovou‐se que basófilos podem induzir a secreção da IL‐4 via IgE alérgeno‐dependente, além da IL‐31 independentemente do estímulo da IgE alérgeno‐específica (ativados por TSLP), via macrófagos que assumem fenótipo M2 na DA. Esses macrófagos M2 secretam IL‐31 sob estímulo da TSLP e também da periostina (secretada pelos fibroblastos), o que poderia explicar o prurido residual na DA em pacientes que usam dupilumabe, um anticorpo monoclonal que bloqueia o receptor IL‐4Rα e o receptor IL‐4Rα acoplado ao IL‐13 R.72
O número de macrófagos M2 expressando IL‐31, macrófagos CD68+ e basófilos nas biopsias de pele acometida pela DA relaciona‐se positivamente com a expressão de TSLP e periostina na epiderme da pele afetada em pacientes com DA e com a gravidade da doença.61
Os eosinófilos ativados na DA expressam quantidades elevadas de receptores de histamina tipo 4, através do estímulo da IL‐4 e IL‐13 via sistema JAK/STAT, levando também ao aumento de produção da IL‐31 pelos eosinófilos. A IL‐18 atua nos receptores (IL‐18Rα) hiperexpressos nos eosinófilos nos pacientes com DA, e a histamina liberada por mastócitos e basófilos aumenta a expressão da IL‐18 nos eosinófilos pela ligação da histamina com H2R e H4R, sugerindo função relevante da IL‐18 e da histamina na inflamação eosinofílica na DA.61
Mastócitos produzem diversas citocinas, tais como IL‐1, 2, 3, 5, 6, 7, 8, 9, 13, 16, 17 e IL‐33. A IL‐33 direciona as respostas imunes inflamatórias do tipo 2 na DA. A secreção da IL‐9 torna possível a sobrevida das células T e a ativação cruzada entre mastócitos, e a IL‐13 contribui para inflamação tipo 2, além de estimular fibroblastos na síntese de colágeno e diferenciação de linfócitos B na conversão de síntese de IgE.61,74
As células NK participam na secreção de diferentes citocinas, como IL‐9, IL‐13, IL‐21, IL‐22 e IL‐31, de modo que, particularmente, a IL‐21 promove ativação de células B e T e sua diferenciação, bem como aumento da atividade das células NK.74
Por fim, a imunopatogênese da DA é fundamentada em resposta imune desregulada, de maneira multidimensional e interligada, afetando a resposta a antígenos, a indução da inflamação, a ruptura da homeostase imune, o desarranjo da integridade arquitetural da epiderme e derme, e produzindo um sintoma cardinal que é o prurido.61
Uma síntese da interferência da desregulação imune quanto à barreira cutânea é apresentada na figura 3.75
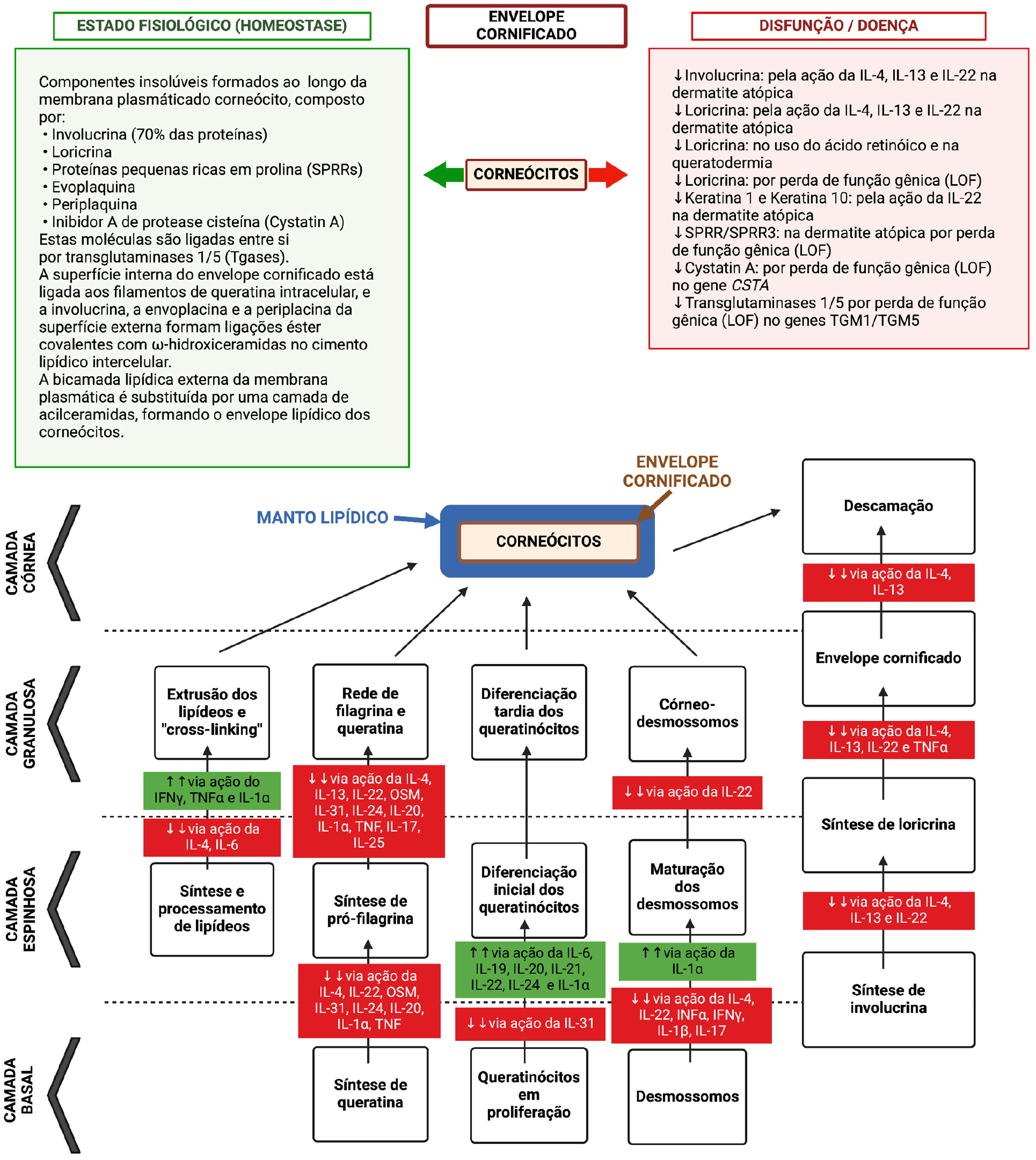
 e supressão de proteínas por ação de citocinas da inflamação atópica. Durante esse processo de diferenciação, o envelope lipídico, a rede de filagrina/queratina e o envelope cornificado são formados, com os desmossomos se diferenciando em corneodesmossomos. Juntos, esses componentes formam uma barreira compacta evitando a permeação de substâncias nocivas ou patógenos na epiderme, bem como irradiação e irritantes. Além disso, essa barreira impede as perdas de água transepidérmica (TEWL) e os solutos associados. O modelo “tijolos e cimento” que ocorre na arquitetura da camada córnea é constituído pelo envelope proteico‐lipídico que envolve o corneócito (proteínas constituídas pela loricrina, involucrina, pequenas proteínas ricas em prolina e filagrina) e o envelope lipídico por uma monocamada de lipídeos, que serve como base para a organização dos lipídeos lamelares intercelulares (compostos por 25% de colesterol, 10%–15% de AGL, 5% de sulfato de colesterol e triacilglicerol e 45%–50% de ceramidas no estrato córneo). Esses lipídeos extracelulares são estocados dentro de corpos lamelares nos queratinócitos da camada espinhosa superior e granulosa, compostos por glucosilceramidas, esfingomielina e fosfolipídeos. O “cimento intercelular” é uma matriz composta por ω‐hidroxiceramidas, colesterol e AGL modificados por enzimas do estrato córneo, além de peptídeos antimicrobianos. Aminoácidos livres resultantes da degradação da filagrina formam a maior parte do denominado “fator de hidratação natural” (NMF) no estrato córneo, representando uma capacidade excepcional de reter água e contribuindo para o pH ácido desta camada. Assim, a matriz lipídeo‐proteica intercorneócitos e os corneócitos ricos em proteínas são fundamentais para a formação da barreira epidérmica funcional. Defeitos genéticos de genes que comandam a síntese dessas proteínas podem concorrer para a dermatite atópica, bem como a supressão da função desses genes por citocinas inflamatórias, como observado neste esquema. O gene da corneodesmosina (CDSN) está suprimido na dermatite atópica, mas a integridade do corneodesmossomo pode também ser modulada negativamente por citocinas tipo 2: IL‐4, IL‐13, IL‐31, IL‐25, IL‐22. IL‐4 promove deficiência de filagrina na DA. A IL‐33 suprime a expressão de claudina nos queratinócitos. A TSLP diminui a síntese de peptídeos antimicrobianos, como a β‐defensina humana via sistema JAK‐STAT e a catelicidina (LL‐37), permitindo maior vulnerabilidade ao eczema herpético e infecções bacterianas.")
Representação do processo de diferenciação dos queratinócitos na epiderme, desde a camada basal até o corneócito, com disfunção da barreira epidérmica pela ação de mutações gênicas com perda de função (LOF) e supressão de proteínas por ação de citocinas da inflamação atópica. Durante esse processo de diferenciação, o envelope lipídico, a rede de filagrina/queratina e o envelope cornificado são formados, com os desmossomos se diferenciando em corneodesmossomos. Juntos, esses componentes formam uma barreira compacta evitando a permeação de substâncias nocivas ou patógenos na epiderme, bem como irradiação e irritantes. Além disso, essa barreira impede as perdas de água transepidérmica (TEWL) e os solutos associados. O modelo “tijolos e cimento” que ocorre na arquitetura da camada córnea é constituído pelo envelope proteico‐lipídico que envolve o corneócito (proteínas constituídas pela loricrina, involucrina, pequenas proteínas ricas em prolina e filagrina) e o envelope lipídico por uma monocamada de lipídeos, que serve como base para a organização dos lipídeos lamelares intercelulares (compostos por 25% de colesterol, 10%–15% de AGL, 5% de sulfato de colesterol e triacilglicerol e 45%–50% de ceramidas no estrato córneo). Esses lipídeos extracelulares são estocados dentro de corpos lamelares nos queratinócitos da camada espinhosa superior e granulosa, compostos por glucosilceramidas, esfingomielina e fosfolipídeos. O “cimento intercelular” é uma matriz composta por ω‐hidroxiceramidas, colesterol e AGL modificados por enzimas do estrato córneo, além de peptídeos antimicrobianos. Aminoácidos livres resultantes da degradação da filagrina formam a maior parte do denominado “fator de hidratação natural” (NMF) no estrato córneo, representando uma capacidade excepcional de reter água e contribuindo para o pH ácido desta camada. Assim, a matriz lipídeo‐proteica intercorneócitos e os corneócitos ricos em proteínas são fundamentais para a formação da barreira epidérmica funcional. Defeitos genéticos de genes que comandam a síntese dessas proteínas podem concorrer para a dermatite atópica, bem como a supressão da função desses genes por citocinas inflamatórias, como observado neste esquema. O gene da corneodesmosina (CDSN) está suprimido na dermatite atópica, mas a integridade do corneodesmossomo pode também ser modulada negativamente por citocinas tipo 2: IL‐4, IL‐13, IL‐31, IL‐25, IL‐22. IL‐4 promove deficiência de filagrina na DA. A IL‐33 suprime a expressão de claudina nos queratinócitos. A TSLP diminui a síntese de peptídeos antimicrobianos, como a β‐defensina humana via sistema JAK‐STAT e a catelicidina (LL‐37), permitindo maior vulnerabilidade ao eczema herpético e infecções bacterianas.
O sistema neurossensorial cutâneo ocupa posição central na patogênese da DA. As terminações nervosas sensoriais estão em contato íntimo com as células residentes e células inflamatórias que infiltram a pele, de modo que elas podem interagir intensamente com essas células e os mediadores liberados tanto nos estágios agudos quanto crônicos da doença. Mediadores inflamatórios da DA podem também sensibilizar nervos sensoriais, induzindo o fenômeno de hiperquinese, ou seja, determinar um aumento de sensibilidade dos nervos aos estímulos pruridogênicos e de aloquinese (estímulos não pruridogênicos, percebidos como prurido), contribuindo para o prurido crônico na DA.76
A DA apresenta a particularidade de, progressivamente, desenvolver aumento da densidade das fibras neurais sensoriais na pele, ocasionando um estado de “sensibilização neural”, propiciando maior reação e interação com o ambiente cutâneo. O aumento da concentração de neurotrofinas, liberadas por projeções nervosas e eosinófilos cutâneos, atuam de maneira sinérgica amplificando a ramificação das terminações nervosas na pele, resultando na hiperinervação da pele inflamada na DA. Essa hiperinervação pode, eventualmente, diminuir o limiar de indução do prurido (hiperquinese) e favorecer a indução do prurido por estímulos não pruridogênicos (aloquinese).76
As fibras neurais sensoriais do tipo C não mielinizadas e de condução lenta constituem as principais vias condutoras ao sistema nervoso central do prurido cutâneo. Basicamente, há duas vias condutoras do prurido: as histaminérgicas (histamina‐sensíveis) e não histaminérgicas (histamina‐insensíveis) inseridas anatomicamente dentro das fibras tipo C da rede neuronal sensorial cutânea. Medicamentos anti‐histamínicos demonstram ter efeito mínimo ou ausência de efeito no controle do prurido na DA, tendo apenas efeitos soníferos nos pacientes. Isso há décadas já foi observado, indicando que a histamina exerce apenas função mínima no prurido associado à DA, ao menos via estímulo dos receptores H1. Além disso, o bloqueio de receptores H4 teve leve efeito antipruriginoso em diferentes estudos experimentais e clínicos.76
Assim, o prurido na DA é primariamente percebido por vias não histaminérgicas nos nervos sensoriais cutâneos, de modo que mediadores inflamatórios pivotais na patogênese da DA podem estimular as vias não histaminérgica e induzir o prurido na DA. Irritantes, alérgenos e produtos bacterianos geram enzimas proteolíticas, as quais ativam os receptores PAR2, pela clivagem proteolítica da porção N‐terminal extracelular desse receptor. Receptores PAR2 estão localizados nos queratinócitos e nervos sensoriais, de modo que atualmente acredita‐se que sua estimulação seja a via principal para o prurido não histaminérgico na DA e indução de inflamação neurogênica, que resulta na liberação de neuropeptídeos, como substância P e peptídeo liberador do gene da calcitonina (CGRP) pelas terminações nervosas na pele (fluxo neural eferente), que retroalimenta receptores nas células da pele intensificando a inflamação.76
Na pele dos pacientes com DA ocorre exposição a várias enzimas proteolíticas exógenas (derivadas de bactérias ou ácaros da poeira domiciliar) ou de fontes endógenas (triptase, tripsina, quimase, calicreína, especialmente a KLK5), as quais são liberadas pelos queratinócitos ou pelas células imunes no processo inflamatório, enaltecendo a relevância dessa via nas fases iniciais da doença. Na fase inicial da DA, a ativação do PAR2 parece anteceder a liberação de alarminas como a TSLP, e o estímulo do TRPV3 pode induzir a liberação da TSLP. A TSPL, junto com a IL‐33, são alarminas que estimulam a inflamação tipo 2 e têm receptores nas terminações neurais sensoriais da pele contribuindo para o prurido. Assim, os queratinócitos transformam estímulos exógenos e endógenos em sinais de prurido via PAR2 e liberam mediadores como a TSLP, criando a chamada “comunicação neuro‐epidérmica” na DA. As terminações nervosas, por sua vez, em resposta, liberam substância P e CGPR. A substância P tem receptores nos próprios nervos, nos queratinócitos e nas células inflamatórias (linfócitos, mastócitos, eosinófilos e basófilos), via receptor 1 de alta afinidade neurocinina e receptores acoplados a proteína G relacionado ao Mas (MrgprX2). A ligação da substância P ao MrgprX2 induz degranulação mastocitária, liberando agentes pruridogênicos como leucotrienos, prostaglandinas, histamina, TNF‐α, proteases e NGF.76
O CGRP é um neuropeptídeo que estimula os nervos sensoriais, vasos sanguíneos e células imunes (células dendríticas e linfócitos T), infiltrando células inflamatórias na pele e propagando a resposta imune Th2, liberando IL‐13 de células T CLA+ na DA. Desse modo, o reflexo neural eferente (antidrômico ou condução oposta do estímulo) de inflamação neurogênica com liberação local de neuropeptídeos antecede a ativação das respostas imunes dos sistemas inato e adaptativo em relação ao prurido.76
Já quando a imunidade adaptativa tipo Th2 se estabelece, a síntese da IL‐4 e IL‐13 ocupa papel central na resposta inflamatória tipo 2 na DA, com múltiplos efeitos nas células epidérmicas, dérmicas e fibras neurais sensoriais. A IL‐4 e a IL‐13 sensibilizam as fibras C sensoriais condutoras do prurido a diminuir o limiar de percepção de estímulos sensitivos a outros estímulos pruridogênicos, como à histamina, IL‐31 e TSLP. IL‐4 e IL‐13 suprimindo a produção epidérmica de filagrina, loricrina e involucrina causam a liberação de enzimas proteolíticas na epiderme que estimulam receptores PAR‐2 e liberam as alarminas (IL25, TSLP, IL‐33), bem como a expressão seletiva de calicreína (KLK0‐7) nos queratinócitos humanos normais, alimentando a inflamação e o prurido. Essas citocinas também amplificam a resposta imune, pois têm receptores em outros linfócitos, mastócitos, basófilos e eosinófilos, desencadeando a liberação de novos mediadores inflamatórios, ou já pré‐formados (histamina, triptase, endotelina‐1, eotaxina, IL‐31), alimentando o prurido crônico da DA. A IL‐31 exerce especial efeito na estimulação do prurido na DA (citocina do prurido), e tem seu receptor nos nervos sensoriais induzindo prurido, o qual na porção de cadeia alfa do receptor da IL‐31 (IL‐31Ra) é conjugado ao receptor de cadeia beta Oncostatin M. A estimulação de ambos os receptores (IL31Ra e OSMbeta) permite a maior densidade e ramificação das terminações neurais sensoriais na pele e aumenta a sensibilidade ao estímulo pela IL‐31 e outros pruritógenos. O processo de sensibilização neural pela IL‐31 contribui para o prurido crônico na DA e tem função crítica no denominado “ciclo prurido‐coçadura”, fenômeno que promove intensamente o desenvolvimento de lesões papulosas prurigoides – isto é, tipo prurigo nodular.72,76,77
Um esquema dos estímulos pruritogênicos da DA está disposto na figura 4.
 se ligam a receptores específicos na membrana dos nervos sensoriais tipo C não mielinizados de condução lenta, respectivamente o receptor neurocinina‐1, NK‐1; receptor muscarínico, M3; receptor da endotelina 1, ETA (endotelina A receptor); receptor da TSLP, TSLPR; receptor STL2da IL‐33. A TSLP produzida por queratinócitos sob estresse induz macrófagos a diferenciarem‐se em fenótipo M2, os quais contribuem para a produção de IL‐31. Macrófagos não diferenciados produzem TNF‐α, que se liga a seu receptor TNFR, anandamide (AEA) que se liga aos receptores canabioides 1/2 (CB1/CB2), encefalina, ENK, a qual se liga aos receptores opioides kappa (KOR) e mu (MOR), além de produzirem o fator de crescimento de nervo (NGF) o qual se liga ao receptor tropomiosina quinase A (TrkA). Eosinófilos produzem leucotrienos (LT), que se ligam ao receptor LTR, NGF e proteases que se ligam ao receptor de protease (PAR2/PAR4). Mastócitos liberam serotonina (5‐hidroxi‐triptofano, 5‐HT), ativando o seu receptor 5HTR, histamina que se liga aos receptores H1/H4, triptase que se liga ao receptor PAR2/4, e substância P. Basófilos são fonte produtora também de histamina e citocinas da inflamação tipo 2, como a IL‐31, IL‐4 e IL‐13, que se ligam aos seus respectivos receptores nas terminações nervosas, os quais têm como via de sinalização intracelular o sistema de enzimas JAK‐STAT. Linfócitos Th2 produzem as mesmas citocinas e os Th22 a IL‐22 que também tem seu receptor específico nas terminações nervosas sensoriais. Todos em conjunto trabalham para a produção do prurido agudo e crônico na dermatite atópica. As fibras neurais sensoriais possuem receptores de potencial transitório (TRP) V1 (receptor de potencial transitório valinoide 1) e TRPA1 (receptor de potencial transitório ankyrin 1), os quais são canais catiônicos inespecíficos. Uma vez que as terminações nervosas tenham sido estimuladas pelas citocinas IL‐4, IL‐13, Il‐22, IL‐33, IL‐31 e seus receptores específicos, a ativação do TRVP1 e/ou TRPA1 induz influxo de cálcio, o qual eventualmente induz a liberação de potenciais de ação via canais de sódio Nav1.7 e Nav1.8 ou Nav1.9. O TRPV1 e TRPA1 devem estar presentes para estes pruritógenos induzirem prurido ou sensibilizar os nervos sensoriais a outros pruritógenos.")
Vias do prurido, seus mediadores e receptores na dermatite atópica. Agentes prurigogênicos produzidos pelos queratinócitos (substância P, SP; acetilcolina, ACOL; endotelina‐1, ET‐1; e alarminas, como a linfopoietina do estroma tímico, TSLP e a IL‐33) se ligam a receptores específicos na membrana dos nervos sensoriais tipo C não mielinizados de condução lenta, respectivamente o receptor neurocinina‐1, NK‐1; receptor muscarínico, M3; receptor da endotelina 1, ETA (endotelina A receptor); receptor da TSLP, TSLPR; receptor STL2da IL‐33. A TSLP produzida por queratinócitos sob estresse induz macrófagos a diferenciarem‐se em fenótipo M2, os quais contribuem para a produção de IL‐31. Macrófagos não diferenciados produzem TNF‐α, que se liga a seu receptor TNFR, anandamide (AEA) que se liga aos receptores canabioides 1/2 (CB1/CB2), encefalina, ENK, a qual se liga aos receptores opioides kappa (KOR) e mu (MOR), além de produzirem o fator de crescimento de nervo (NGF) o qual se liga ao receptor tropomiosina quinase A (TrkA). Eosinófilos produzem leucotrienos (LT), que se ligam ao receptor LTR, NGF e proteases que se ligam ao receptor de protease (PAR2/PAR4). Mastócitos liberam serotonina (5‐hidroxi‐triptofano, 5‐HT), ativando o seu receptor 5HTR, histamina que se liga aos receptores H1/H4, triptase que se liga ao receptor PAR2/4, e substância P. Basófilos são fonte produtora também de histamina e citocinas da inflamação tipo 2, como a IL‐31, IL‐4 e IL‐13, que se ligam aos seus respectivos receptores nas terminações nervosas, os quais têm como via de sinalização intracelular o sistema de enzimas JAK‐STAT. Linfócitos Th2 produzem as mesmas citocinas e os Th22 a IL‐22 que também tem seu receptor específico nas terminações nervosas sensoriais. Todos em conjunto trabalham para a produção do prurido agudo e crônico na dermatite atópica. As fibras neurais sensoriais possuem receptores de potencial transitório (TRP) V1 (receptor de potencial transitório valinoide 1) e TRPA1 (receptor de potencial transitório ankyrin 1), os quais são canais catiônicos inespecíficos. Uma vez que as terminações nervosas tenham sido estimuladas pelas citocinas IL‐4, IL‐13, Il‐22, IL‐33, IL‐31 e seus receptores específicos, a ativação do TRVP1 e/ou TRPA1 induz influxo de cálcio, o qual eventualmente induz a liberação de potenciais de ação via canais de sódio Nav1.7 e Nav1.8 ou Nav1.9. O TRPV1 e TRPA1 devem estar presentes para estes pruritógenos induzirem prurido ou sensibilizar os nervos sensoriais a outros pruritógenos.
Na apresentação clínica da doença, esses elementos se manifestam em sua morfologia como lesões agudas da DA compreendendo eritema, edema e exsudação, e as lesões crônicas xerose, liquenificação e hiperpigmentação.74
Inicialmente, a fase inicial da DA foi atribuída a uma resposta direcionada Th2, enquanto a fase crônica era marcada pela predominância das respostas Th1.74 Além disso, as lesões agudas na DA revelam infiltrado linfocitário abundante na pele, bem como expressão acentuada de IL‐4, IL‐5, IL‐13, IL‐31 e IL‐33, característicos das respostas Th2.61,74
A despeito disso, estudos posteriores mostraram que a resposta Th2 é acompanhada simultaneamente da ativação Th22 e uma indução em menor proporção de marcadores de ativação de resposta Th17.74 Desse modo, a atual compreensão das formas crônicas cutâneas de lesões da DA demonstra aumento simultâneo e retroalimentado dos eixos de citocinas Th2 e Th22, e adicionalmente aumento na expressão de respostas Th1, mas não marcadores específicos da resposta Th17.74
Essas características podem ser relevantes quanto à elaboração da estratégia terapêutica e na compreensão dos perfis de desequilíbrio da resposta imune subjacentes. Por exemplo, considerando a etnicidade e apresentação clínica da DA, o papel do eixo Th17 tem maior participação na fase crônica da DA, e em maior proporção nas formas de DA intrínseca, nas crianças e nos pacientes de etnia asiática, em que agentes inibidores da JAK1 e/ou imunobiológicos anti‐IL17, anti‐IL‐23, anti‐IL‐22 podem, no futuro, demonstrar‐se alternativa aos agentes de primeira linha terapêutica ou nos casos refratários. Da mesma maneira, estudos de fase III em condução com anticorpos monoclonais com alvo nas interações nas moléculas coestimuladoras OX40L‐OX40 nas interações entre células apresentadoras de antígenos e linfócitos T podem ocupar papel relevante, quando se pretende ação terapêutica que envolva vários eixos imunes ativos, ou mesmo em populações com elevado índice de miscigenação racial.78
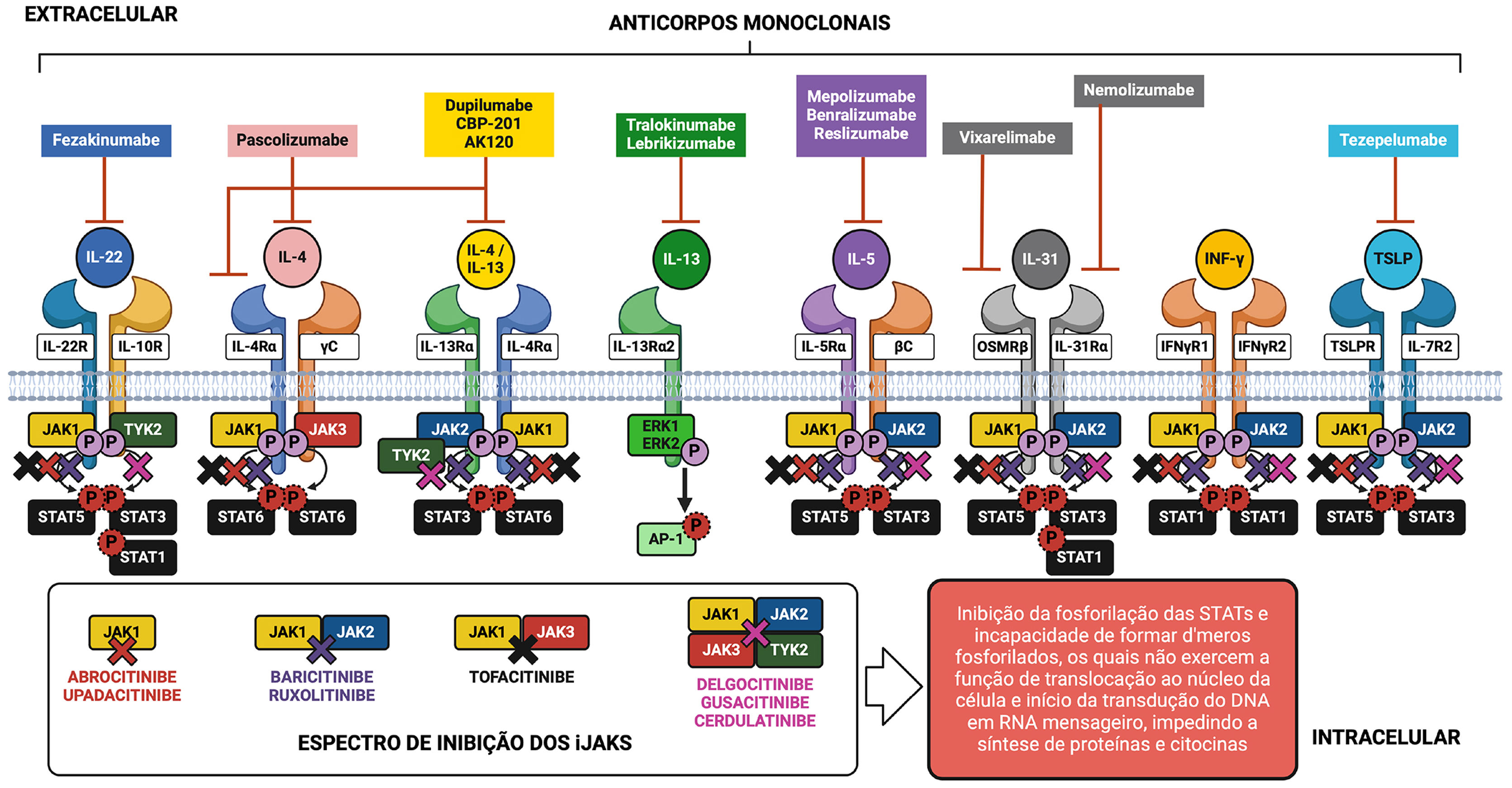
O conhecimento sobre a ação de citocinas por meio de seus receptores celulares específicos e a existência do sistema de sinalização intracelular JAK‐STAT na DA propiciou o surgimento de estratégias terapêuticas direcionadas ao bloqueio de citocinas ou de seus receptores extracelulares com anticorpos monoclonais, de uso subcutâneo e também a abordagem de interrupção parcial do sistema JAK‐STAT por meio de fármacos denominados pequenas moléculas de uso oral, como demonstrado na figura 5.
Base patogênica dos fenótipos de dermatite atópica
O polimorfismo das apresentações clínicas da DA é subsidiado por base genética, níveis de comprometimento da barreira cutânea e por padrões de desbalanço da resposta imune. As tabelas 3 e 4 resumem os diferentes aspectos fenotípicos da DA e sua relação com níveis séricos da IgE, além de padrões de respostas imunes mais ativas em diferentes grupos étnicos. O reconhecimento desses aspectos fenotípicos e de sua base fisiopatológica subjacente pode também orientar a melhor opção terapêutica.
Principais características dos fenótipos de DA associados aos níveis de IgE
| Características | Extrínseca | Intrínseca |
|---|---|---|
| IgE sérica | ↑↑↑ | Normal |
| Frequência | 70%–80% | 20%–30% |
| Início da DA | Infância > Adulto | Adulto > Infância |
| Mutação da filagrina | ↑↑↑ | ↑ |
| Ictiose vulgar | ↑↑↑ | ↑ |
| Hiperlinearidade palmar | ↑↑↑ | ↑ |
| Hipersensibilidade | Asma/rinite, alergia alimentar | Dermatite de contato a metais |
| Infecções cutâneas | ↑↑↑ | ↑ |
| Resposta preponderante | Th2: IL‐4, IL‐13, IL‐31 | Th22: IL‐22; Th17: IL‐17 |
| Marcadores de gravidade | Citocinas Th2 (IL‐4) | Citocinas Th17 (IL‐17) |
| Eosinofilia dérmica | ↑↑↑ | ↑ |
| Predominância clínica | Eczema subagudo | Liquenificação/prurigo |
Principais características ligadas à etnia dos doentes com dermatite atópica
| Etnia | Africana/negra | Asiática (japoneses, chineses/han, coreanos) | Europeia |
|---|---|---|---|
| Principais alterações genéticas | FLG‐2 (LOF), TCHH, TCHHL1, CLDN1, HRNR, CRNN, IL4, IL4RA, IL7R, IRF2, TSLP | FLG null, FCER1A, DEFB1, IL‐4, IL‐13, IL‐31, IL‐5, IL‐12B, IL‐18, IL‐18RAP, IL‐13RA1 TLR2, SPINK5 | FLG (LOF), FCER1A, DEFB1, IL7R, IL‐4, IL‐13, IL‐31, IL‐4RA, IL‐13RA1 TLR2, IRF2, TSLPR |
| Polarização imunológica | Th2 ↑↑↑, Th22↑↑, Th17 (+/−), Th1 (+/−) | Th22↑↑↑↑, Th2 ↑↑↑, Th17↑↑, Th1↑ | Th2 ↑↑↑, Th22↑↑↑, Th1↑↑, Th17↑ |
| Predominância clínica | Liquenificação | Lesões circunscritas+liquenificação | Eczema extenso+malar (infância) |
| Implicação terapêutica | Estratégias anti‐Th2/Th22 | Estratégia de ação ampla (imunossupressores e inibidores da JAK) | Estratégias anti‐Th2/Th22 |
FLG, filagrina; ILC2, célula linfoide tipo 2da imunidade inata; LOF, loss‐of‐function (perda de função); IL, interleucina; INF, interferona; IRF2, fator regulador 2 do Intérferon; TNF, fator de necrose tumoral; TRL2, Toll‐like receptor 2; TSLP, linfopoietina do estroma tímico; FCER1A, receptor de alta afinidade da IgE subunidade alfa; DEF1, beta‐defensina1; SPINK5, inibidor da serina peptidase kazal tipo 5; HRNR, hornerina; CLDN1, claudina 1; Th, linfócito T‐helper; JAK, janus kinase; CRNN, cornulina.
Cerca de 70%–80% dos adultos com DA demonstram elevados níveis séricos de IgE e IgEs específicas a vários alérgenos (fenótipo extrínseco, alérgico ou exógeno), no qual a gravidade das lesões cutâneas se correlaciona com os níveis séricos de IgE. Ainda, foi relatada melhora clínica nas lesões eczematosas em ambientes livres do alérgeno com IgE específica. Apesar disso, omalizumabe não é eficaz no tratamento do eczema da DA extrínseca, e a DA não deve ser justificada apenas pela ocorrência de IgE sérica elevada. Entretanto, a presença de IgE específicas a determinados alérgenos exerce um papel no desenvolvimento da dermatite eczematosa decorrente da hipersensibilidade tipo tardia IgE‐mediada, funcionando primordialmente como amplificadora de outros fatores envolvidos na patogênese da DA.79,80
Por outro lado, na DA intrínseca (endógena, atopiforme, ou não IgE‐alérgica) não é detectada sensibilização a alérgenos ambientais, e os níveis séricos de IgE são normais. Em vez disso, a inflamação cutânea é muitas vezes atribuída a fatores como barreira cutânea comprometida, desequilíbrios na microbiota da pele e disfunções do sistema imunológico. A DA intrínseca tende a ser mais persistente e menos responsiva aos tratamentos convencionais. Esse fenótipo pode não preencher os critérios diagnósticos habituais de DA (p. ex.; Hanifin e Rajka), levando ao atraso diagnóstico e terapêuticas inadequadas.81
Alguns pacientes, ainda, podem apresentar sensação intensa e crônica de prurido, conhecida como prurido neuropático. Esse fenótipo está associado a anormalidades na via nervosa periférica e central; o menor limiar de prurido e a coçadura recorrente podem contribuir para a persistência da inflamação na DA.
É importante notar que os fenótipos intrínseco e extrínseco não são mutuamente exclusivos, e alguns pacientes podem exibir características de ambos os fenótipos ou migrar da predominância de um para o outro durante o curso da doença. O reconhecimento dessas distinções pode ser útil para orientar o manejo terapêutico, especialmente no que diz respeito à identificação de gatilhos ambientais.
Morfologicamente, de acordo com o padrão de lesão cutânea, uma rede de citocinas diferentes assume o protagonismo da resposta inflamatória, o que tem implicação terapêutica. No eczema agudo, as lesões são subsidiadas por expressão tecidual de IL‐1α e IL‐1β, IL‐33, IL‐4, IL‐13, e IL‐22. No eczema subagudo, as principais citocinas são IL‐4, IL‐13, IL‐25, IL‐31, IL‐33 e TLSP. Já no eczema crônico, há predominância de citocinas Th1 e Th17: IFNγ, TNFα, IL‐17A e IL‐17F.82
Por fim, casuísticas de diferentes grupos étnicos originais também compartilham fenótipos ligados ao protagonismo dos componentes patogênicos da DA (tabela 4). Entretanto, a globalização e miscigenação populacional, como acontece no Brasil, tende a diminuir a preponderância de fenótipos ligados às etnias na apresentação clínica da DA.83
Microbioma na dermatite atópicaA pele humana é um ecossistema rico habitado por uma diversidade de microrganismos, incluindo bactérias, fungos e vírus. As bactérias dos filos Actinobacteria, Firmicutes e Proteobacteria são os mais prevalentes. Fungos, como Malassezia spp., e ácaros Demodex spp., também são componentes importantes da microbiota cutânea. Esses microrganismos, principalmente comensais, interagem diretamente com o hospedeiro, agem como proteção contra patógenos, estimulam a barreira cutânea e regulam a resposta imune; entretanto, seu desequilíbrio pode contribuir para a exacerbação da DA.84–86A composição da microbiota da pele se inicia ao nascimento, diferindo entre recém‐nascidos de parto vaginal e cesariana. Essa composição é influenciada por diversos fatores ao longo da vida, incluindo mudanças no pH da pele, umidade, poluição e produção de sebo.87–89 Alterações no microbioma podem preceder o desenvolvimento de DA, com a puberdade marcando outra fase de mudança significante em virtude da influência de hormônios sexuais.90–93A microbiota cutânea participa na manutenção da função de barreira da pele, influenciando a diferenciação e regeneração celular. Por exemplo, estafilococos coagulase‐negativos, como S. hominis, produzem substâncias que inibem o crescimento de S. aureus.94–96Pacientes com DA frequentemente exibem disbiose, manifestada pela diminuição na diversidade microbiana, com um ambiente de pele que favorece o crescimento de S. aureus, ligado a um aumento no pH cutâneo e deficiências em componentes antimicrobianos.97–102A flora cutânea saudável é necessária para manter uma barreira microbiana, pois certas bactérias na pele sã suprimem bactérias virulentas e induzem a expressão de peptídeos antimicrobianos (AMPs), controlando a inflamação. O pH da pele ajuda a determinar as populações microbianas da pele, e um pH aumentado pode aumentar a suscetibilidade a infecções. A flora saudável, juntamente com a expressão normal de proteínas cutâneas, enzimas, proteases e equilíbrio de citocinas inflamatórias, são cruciais para manter um pH ácido normal da pele.99
A infecção por S. aureus inibe a expressão de fatores de diferenciação tardia nos queratinócitos, como filagrina, loricrina, queratina 1 e 10 e desmocolina 1, o que pode comprometer ainda mais a barreira cutânea. Além disso, componentes bacterianos de S. aureus estão associados à composição alterada dos lipídios intercelulares.103,104
O tratamento de condições cutâneas com antibióticos e agentes antibacterianos pode ser eficaz; contudo, seu uso prolongado aumenta o risco de promover resistência bacteriana. Terapias recentes, como dupilumabe e tralokinumabe, favorecem o reequilíbrio da microbiota da pele em pacientes com DA.94,105 O emprego de probióticos, pré‐bióticos e pós‐bióticos é também explorado para combater a disbiose cutânea.95
Há menor volume de informações quanto à microbiota intestinal e DA. O intestino humano hospeda mais de 100 trilhões de microrganismos e é influenciado por fatores como genética, dieta e tratamentos médicos. A composição da microbiota intestinal afeta o desenvolvimento imunológico e distúrbios podem levar a condições como doenças inflamatórias e obesidade. A disbiose na microbiota intestinal está ligada à DA, caracterizada pela diminuição da diversidade microbiana e aumento da presença de bactérias nocivas como o Clostridium difficile. Esse desequilíbrio pode levar ao aumento da permeabilidade intestinal, com disfunção das zonas de oclusão interepiteliais no intestino, permitindo que as toxinas bacterianas permeiem o epitélio e impactem a resposta imune local e sistêmica, inclusive na pele.106 Fatores dietéticos, particularmente a dieta ocidental, contribuem para a disbiose intestinal e DA, alterando os genes microbianos do intestino e as respostas inflamatórias do organismo.107
Aspectos neuropsiquiátricosA DA impacta a qualidade de vida dos pacientes e de suas famílias, cuja contingência crônica manifesta‐se, neuropsicologicamente, de várias maneiras, incluindo ansiedade, depressão, transtornos de comportamento disruptivo, transtornos do desenvolvimento neurológico, de déficit de atenção/hiperatividade (TDAH), do espectro autista e ideação suicida, sendo essas associações especialmente marcantes em casos graves de DA.108–110A prevalência de TDAH em crianças com DA foi estimada em 7,1% (95% IC 5,4–8,9%), enquanto em crianças sem DA foi de 4,1%, em uma casuística israelense.111 Essas crianças apresentam problemas no sono em taxas mais elevadas do que aquelas com apenas DA. A privação de sono nos primeiros 3 anos de vida está ligada a hiperatividade e menor desempenho cognitivo aos 6 anos, evidenciando que o sono insuficiente pode ter efeitos duradouros no desenvolvimento neurocognitivo.112 Mães de bebês com DA reportam sentimentos de depressão e ansiedade mais acentuados em comparação com mães de crianças sem DA. Além disso, pais de crianças com DA relatam que o sono interrompido está diretamente relacionado com seus próprios níveis de ansiedade e depressão.113,114
Um estudo longitudinal que acompanhou 1.578 indivíduos do nascimento até os 10 anos de idade mostrou que a DA na infância e problemas de sono são preditores significantes de problemas de conduta e emocionais aos 10 anos.115 A pesquisa também associa o TDAH ao uso de anti‐histamínicos (OR=1,88; 95% IC 1,04–3,39), embora estudos sobre cetirizina não tenham encontrado diferenças significantes em comparação com placebo nas avaliações de comportamento e desenvolvimento das crianças.116,117
Além disso, a presença e a intensidade do prurido estão positivamente correlacionadas com sintomas de depressão, ansiedade e estresse ao longo da vida. A elevada incidência de sintomas de saúde mental em crianças com DA pode ser parcialmente decorrente da coexistência de outras condições. Estudos mostram que comorbidades atópicas estão associadas a maiores prevalências de várias condições psicológicas, como autismo e TDAH.110
Adolescentes com DA relatam altas taxas de vergonha, evitam atividades sociais e têm menos amigos, enquanto crianças com DA sofrem de bullying e têm mais experiências negativas com colegas e professores.118,119 A DA leve a moderada está associada a um aumento de 29% a 84% no risco de comportamento internalizante dos 4 aos 16 anos, embora não esteja diretamente ligada a um aumento no risco de depressão dos 9 aos 18 anos.120 Além disso, tais comportamentos na infância estão relacionados à depressão e ansiedade na idade adulta.121
Uma revisão sistemática indicou que aproximadamente uma em cada seis pessoas com DA apresenta depressão clínica, uma em quatro apresenta sintomas depressivos e uma em oito tem ideação suicida.122 Pacientes com DA têm maiores chances de depressão clínica e sintomas depressivos comparados a indivíduos saudáveis, entretanto, semelhantemente a pessoas com outros distúrbios dermatológicos crônicos. Finalmente, quase um terço dos pais de crianças com DA estão deprimidos, e há uma prevalência elevada de ideação suicida entre pacientes com DA.122
O tratamento farmacológico da DA reduz escores de ansiedade e depressão nos pacientes com doença moderada a grave, enquanto intervenções não farmacológicas levam à redução nos escores de ansiedade.123 Ainda, intervenções psicológicas levaram à melhora da qualidade de vida, redução dos escores de gravidade do eczema e do prurido.124
Identificou‐se associação entre DA e outras alterações neuropsiquiátricas, incluindo autismo (OR=2,14; 95% IC 1,39–3,29), TDAH (OR=1,78; 95% IC 1,21–2,62), distúrbios de personalidade, comportamentais e esquizofrenia (OR=1,45; 95% IC 1,23–1,72),125 além de uma nova associação entre DA e risco aumentado de desenvolver demência (OR=2,02; 95% IC 1,24–3,29).126 Isso destaca a complexidade e a gravidade do impacto da DA na saúde mental e na qualidade de vida dos pacientes e suas famílias.
Embora a relação entre DA e condições neuropsiquiátricas seja destacada, a causa subjacente dessas associações não é totalmente conhecida, assim como a interação entre os sintomas dermatológicos (p. ex., prurido) e o agravamento dos sintomas neuropsiquiátricos. A melhor compreensão dessa dinâmica deve levar a estratégias de prevenção primária e secundária para reduzir o impacto neuropsiquiátrico da DA, como intervenções precoces em pacientes de alto risco e medidas para melhorar a qualidade de vida dos pacientes e suas famílias.
Comorbidades em adultos e idosos com DAO estado inflamatório crônico pode propiciar eventos cardiovasculares maiores (angina, infarto aguda do miocárdio, arritmias cardíacas etc.), insuficiência cardíaca, tromboembolismo venoso e malignidades linfoproliferativas nos doentes com DA.27
Em um estudo de coorte na população do Reino Unido, que avaliou 625.083 adultos com DA em seguimento ao menos por cinco anos, sem receber terapia imunossupressora, comparados a outros 2.678.888 adultos sem DA, pareados por sexo e idade, observou‐se que entre adultos com DA grave (10.543 pacientes) houve risco 1,86 vez maior de linfoma não Hodgkin e risco 21,05 vezes maior de linfoma cutâneo de células T.127
Dados de atendimento de doentes no Reino Unido entre 1994 e 2025 demonstraram que adultos com DA grave apresentam maior risco de infarto do miocárdio Hazard Ratio razão de risco instantâneo [HR=1,27; 95% IC 1,15–1,39]), acidente vascular encefálico (HR=1,21; 95% IC 1,13–1,30) e embolia pulmonar (HR=1,39; 95% IC 1,21–1,60), quando comparados a adultos sem DA.128
Apesar da sabida associação entre comorbidades sistêmicas e DA, e a importância da identificação dessas condições, não se conhece ainda a relação causal direta da inflamação crônica da DA com tais agravos, tampouco se o tratamento eficaz da DA reduz a incidência dessas comorbidades.
Considerações finaisDA é uma condição complexa, multifatorial crônica e recorrente que apresenta fenótipos variáveis resultantes da interação entre a base genética, defeito de barreira cutânea, dos perfis de resposta imune e reatividade à exposição ambiental. A melhor compreensão da sua patogênese leva à possibilidade de intervenção nos fatores preponderantes para o agravamento dos quadros, em cada circunstância, assim como para promover a redução da atividade basal da doença, em casos subclínicos, prolongando os períodos de remissão.
Suporte financeiroNenhum.
Contribuição dos autoresPaulo Ricardo Criado: Idealização do estudo, escrita e aprovação do texto final.
Roberta Fachini Jardim Criado: Idealização do estudo, escrita e aprovação do texto final.
Mayra Ianhez: Idealização do estudo, escrita e aprovação do texto final.
Hélio Amante Miot: Idealização do estudo, escrita e aprovação do texto final.
Caio César Silva de Castro: Idealização do estudo, escrita e aprovação do texto final.
Roberto Bueno‐Filho: Idealização do estudo, escrita e aprovação do texto final.
Conflito de interessesDr. Paulo Criado: Advisory board – Pfizer, Galderma, Takeda, Hypera, Novartis, Sanofi; Pesquisa clínica – Pfizer, Novartis, Sanofi, Amgen e Lilly; Palestrantre – Pfizer, Abbvie, Sanofi‐Genzyme, Hypera, Takeda, Novartis.
Dra. Roberta Fachini Jardim Criado: Advisory board – Pfizer, Takeda, Hypera, Novartis, Sanofi; Pesquisa clínica – Pfizer, Novartis, Sanofi e Lilly; Palestrantre – Pfizer, Abbvie, Sanofi‐Genzyme, Hypera, Takeda, Novartis.
Dra. Mayra Ianhez: Advisory board – Galderma, Sanofi, Pfizer, Novartis, Abbvie, Janssen, UCB‐Biopharma, Boehringer‐Ingelheim; Palestrante – Galderma, Sanofi, Pfizer, Theraskin, Novartis, Abbvie, Janssen, Leopharma, FQM.
Dr. Caio Castro: Advisory board – Sanofi, Aché, Sun‐Pharma, Galderma; Palestrante – Abbvie, Jansen, Novartis, Sanofi, Leo‐ Pharma; Pesquisa clínica – Abbvie, Pfizer, Jansen, Sanofi.
Dr. Roberto Bueno‐Filho: Advisory board – Janssen, Novartis; Pesquisa clínica – Sanofi, Janssen e Lilly; Palestrantre – Pfizer, Janssen, Sanofi, Novartis.
Dr. Hélio Miot: Advisory Board – Johnson & Johnson, L’Oréal, Theraskin, Sanofi e Pfizer; Pesquisa clínica – Abbvie, Pierre‐Fabre, Galderma e Merz.
Como citar este artigo: Criado PR, Miot HA, Bueno‐Filho R, Ianhez M, Criado RF, Silva de Castro CC. Update on the pathogenesis of atopic dermatitis. An Bras Dermatol. 2024;99. https://doi.org/10.1016/j.abd.2024.06.001
Trabalho realizado na Faculdade de Medicina do ABC, Santo André, SP, Brasil.
