






A dermatose purpúrica pigmentada se manifesta clinicamente como máculas hiperpigmentadas e petéquias, predominantemente nas extremidades inferiores. À histopatologia, é caracterizada por um infiltrado linfocítico na derme superior, eritrócitos extravasados e depósitos de hemossiderina. Existe uma variante incomum, conhecida como dermatose purpúrica pigmentada granulomatosa, que se caracteriza histologicamente pela presença de granulomas não necrotizantes associados aos achados clássicos de outras dermatoses purpúricas pigmentadas. Afeta mais frequentemente mulheres de meia‐idade de origem asiática, predominantemente nas extremidades inferiores. É apresentado o caso de uma paciente do sexo feminino com dermatose purpúrica pigmentada granulomatosa nas extremidades inferiores com distribuição blaschkoide.
A dermatose purpúrica pigmentada (DPP) ou capilarite representa um grupo heterogêneo de dermatoses de etiologia desconhecida, que são caracterizadas por máculas pigmentadas do vermelho ao castanho e petéquias, predominantemente nas extremidades inferiores. Classicamente, foram descritas cinco variantes clínicas da DPP: púrpura anular telangiectásica de Majocchi, púrpura pigmentar progressiva de Schamberg, dermatite liquenoide purpúrica e pigmentada de Gougerot e Blum, púrpura eczematoide de Doucas e Kapentanakis e líquen aureus.1 Os achados histológicos clássicos incluem um infiltrado linfocítico perivascular e superficial, extravasamento de eritrócitos e depósitos de hemossiderina, sem vasculite. Uma variante granulomatosa da DPP foi descrita recentemente, cuja principal característica histológica é a presença de granulomas não necrotizantes associados aos achados clássicos da DPP.2
Relata‐se o caso de uma paciente latino‐americana do sexo feminino com máculas hiperpigmentadas nas extremidades inferiores, algumas delas com distribuição blaschkoide, cuja biópsia cutânea revelou a presença de granulomas não necrotizantes juntamente com as características comuns da DPP.
Relato do casoUma paciente do sexo feminino de 62 anos se apresentou com uma história de máculas pigmentadas e assintomáticas em ambas as pernas por seis meses. Ela também tinha história de diabetes mellitus tipo II, hipertensão, dislipidemia e hipotireoidismo e estava sob tratamento com metformina, losartana, atenolol, atorvastatina e levotiroxina. Não havia outros sintomas associados ou alterações na medicação usual.
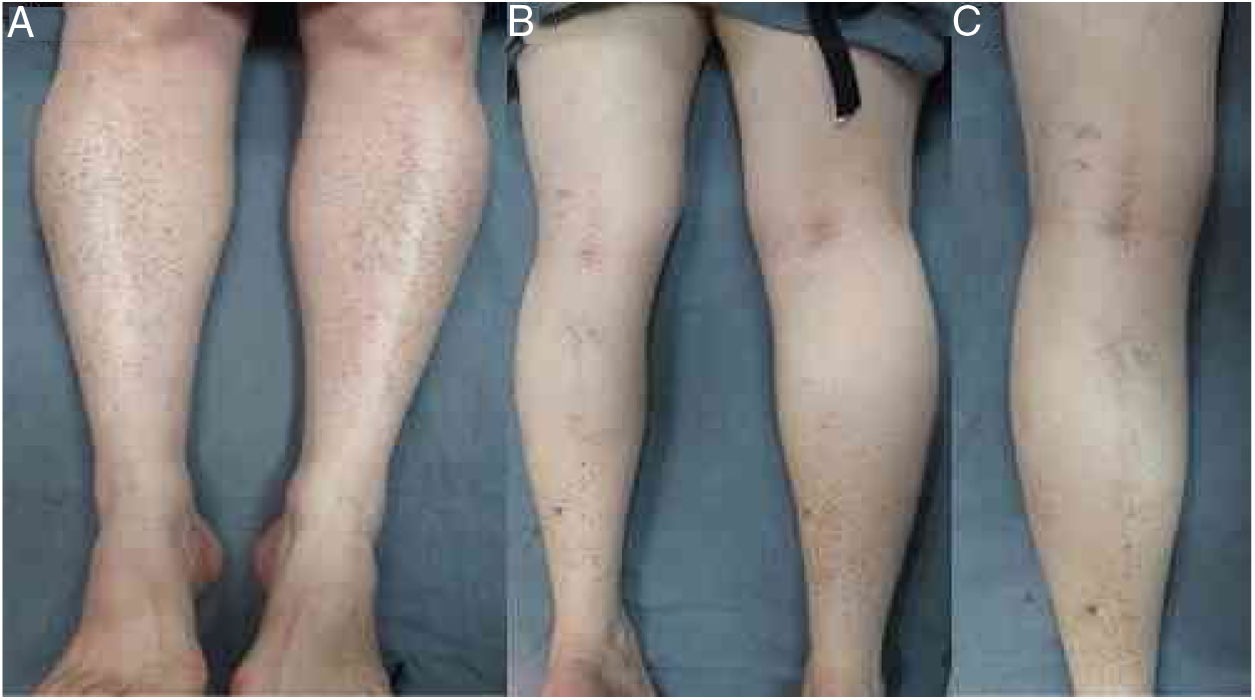
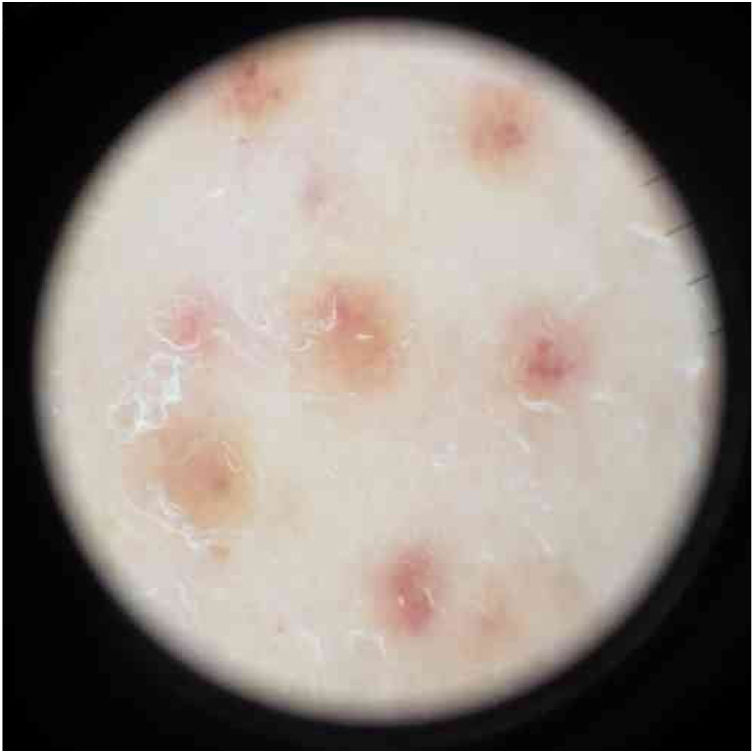
O exame físico revelou pequenas máculas vermelho‐acastanhadas de 2 a 3mm e petéquias não palpáveis distribuídas simetricamente nas extremidades inferiores (fig. 1A). Na face posterior da perna esquerda, as lesões tinham uma distribuição linear, blaschkoide (fig. 1 B e C). A dermatoscopia revelou pontos e glóbulos vermelho‐acastanhados dispostos num fundo de pigmentação vermelho‐acobreada (fig. 2).
A biópsia da pele revelou adelgaçamento da epiderme, infiltrado liquenoide em faixa com linfócitos na derme papilar e reticular (fig. 3A), pequenos granulomas não necrotizantes na derme, formados por depósitos de células epitelioides e células gigantes multinucleadas, com uma pequena coroa linfoplasmocitária e sem necrose (fig. 3B). Foi observado extravasamento de eritrócitos e poucos eosinófilos, mas não havia vasculite. Depósitos de hemossiderina foram evidenciados com a coloração azul da Prússia para ferro (fig. 3C). As colorações de ácido periódico‐Schiff (PAS) e Ziehl‐Neelsen foram negativas.
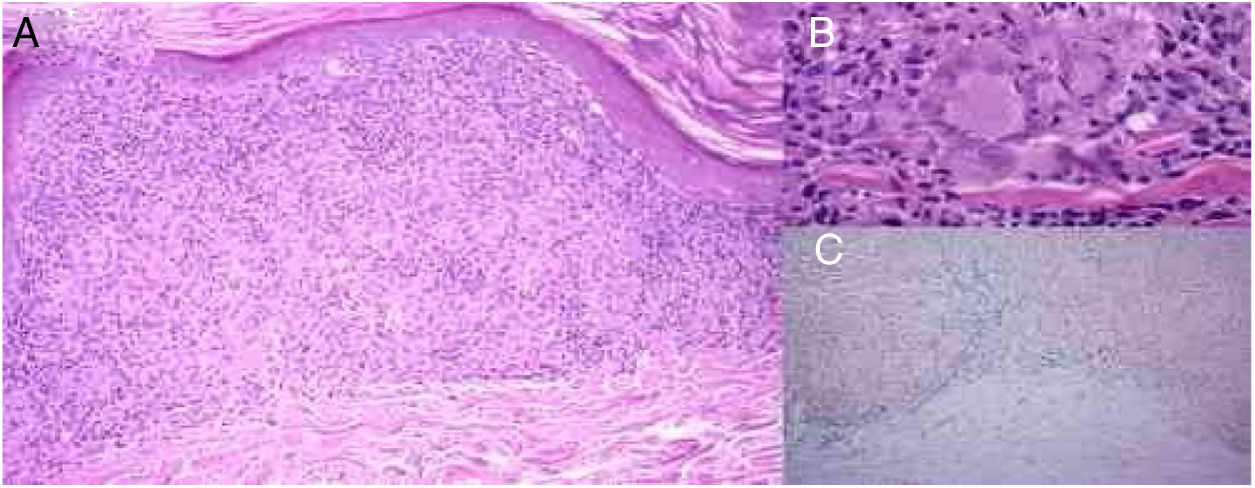
A, Adelgaçamento da epiderme, infiltrado liquenoide com linfócitos e histiócitos que forma granulomas não necrotizantes na derme e extravasamento de eritrócitos (hematoxilina & eosina, 100×). B, Pequenos granulomas não necrotizantes na derme, formados por acúmulos de células epitelioides e células gigantes multinucleadas (hematoxilina & eosina,400×). C, Extravasamento de eritrócitos e depósitos de hemossiderina na derme, especialmente abaixo do infiltrado inflamatório com granulomas (azul da Prússia,100×).
Estudos laboratoriais mostraram colesterol LDL e triglicerídeos elevados. A proteína C‐reativa e a taxa de hemossedimentação estavam normais, sem disfunção orgânica. A história clínica e os achados histológicos confirmaram o diagnóstico de DPP granulomatosa.
DiscussãoA DPP granulomatosa foi descrita pela primeira vez por Saito e Matsuoka em 1996 e foi a última variante identificada da DPP.2 Até o momento, somente 27 casos de DPP granulomatosa foram publicados, com clara predominância no sexo feminino (74%), entre 9 e 75 anos; metade dos casos ocorreu em pacientes asiáticos.3 Recentemente, observou‐se um aumento de relatos de DPP granulomatosa em pacientes brancos, mas não há caso publicado na população latino‐americana.
Clinicamente, a DPP granulomatosa se apresenta como máculas vermelho‐acastanhadas e petéquias, como em outras DPPs; as extremidades inferiores foram a localização mais frequente.4 Os achados dermatoscópicos em neste caso são semelhantes aos de outros relatos, com múltiplos pontos castanho‐avermelhados, arredondados a ovais, glóbulos e máculas em um fundo de pigmentação vermelho‐acobreada sem rede.5 Há relatos de capilarite linear unilateral, líquen aureus e doença de Schamberg com lesões dispostas em distribuição linear.2,6 No entanto, até o momento não há relatos de DPP granulomatosa com distribuição linear ou blaschkoide.
Histologicamente, a DPP granulomatosa apresenta os achados clássicos das outras DPPs, além de um infiltrado linfo‐histiocítico e da formação de granulomas não necrotizantes na derme papilar, alguns dispostos ao redor do plexo vascular.2,3 Outros achados incluem alterações vacuolares ou infiltrado liquenoide na junção dermoepidérmica.4,7 O diagnóstico diferencial histopatológico engloba outras doenças que se apresentam com granulomas: doenças infecciosas, como tuberculose e micobacteriose atípica, e doenças não infecciosas, como sarcoidose, reação medicamentosa, doença de Crohn metastática, vasculite granulomatosa e micose fungoide.5
A etiologia da DPP granulomatosa ainda é desconhecida. Essa variante tem sido associada à hiperlipidemia em mais de 50% dos casos.3 Tem sido postulado que a hiperlipidemia poderia causar inflamação crônica e uma resposta Th1 insuficiente, levando assim à formação do granuloma.5 Outras teorias sustentam que um dano vascular subjacente, induzido pela deposição de lipídios nas células endoteliais, poderia resultar em uma resposta granulomatosa.4
A autoimunidade é um achado comum em pacientes com DPP granulomatosa, relatada em mais da metade dos casos. Os cenários mais frequentes são hipotireoidismo, colite ulcerativa, síndrome de Sjögren e esclerose múltipla.3,8 Alguns casos apresentam marcadores autoimunes positivos, como anticorpos antinucleares, fator reumatoide e crioglobulinas, sugerindo que a autoimunidade também possa ter um papel nessa variante. São necessários mais estudos para estabelecer uma associação.3 Outros achados frequentemente descritos são hipertensão arterial e diabetes mellitus.2
A DPP granulomatosa é uma doença frequentemente assintomática e benigna.2 Assim, o tratamento geralmente é reservado para o controle dos sintomas associados, como prurido, e das alterações estéticas, em pacientes angustiados com a aparência da pele. O tratamento dos fatores desencadeantes suspeitos, como a dislipidemia, e o tratamento da estase venosa associada, quando presente, podem ser benéficos.2,3 Anti‐histamínicos são usados para controlar o prurido – no entanto, eles não têm efeito sobre a evolução da doença. Uma revisão sistemática9 das estratégias terapêuticas para a DPP revelou que corticosteroides locais, inibidores da calcineurina locais, rutosídeo, ácido ascórbico em altas doses, colchicina, pentoxifilina, fototerapia e laserterapia promoveram boas respostas. A ciclosporina A e outros imunossupressores podem ser usados na doença refratária. São necessários estudos sistemáticos maiores para avaliar a eficácia das estratégias terapêuticas.
Suporte financeiroNenhum.
Contribuição dos autoresDaniela Carvajal: aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Claudia Quiroz: aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Claudia Morales: aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Javier Fernández: aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Conflito de interessesNenhum.
Como citar este artigo: Carvajal D, Quiroz C, Morales C, Fernández J. Granulomatous pigmented purpuric dermatosis: report of a Latin‐American case with blaschkoid distribution. An Bras Dermatol. 2019;94:582–5.
Trabalho realizado no Hospital Clínico, Universidad de Chile, Santiago, Chile.
