







A paquidermoperiostose ou osteoartropatia hipertrófica primária é uma doença hereditária rara, caracterizada por baqueteamento digital, paquidermia e periostose. Sua patogênese é incerta e o diagnóstico é firmado com base em dados clínicos e radiológicos. Relata‐se uma forma completa da síndrome em paciente do sexo masculino com quadro iniciado na adolescência, com achados clínicos e radiológicos compatíveis. Apresentam-se os os três achados cardinais, além de outras manifestações associadas, como hiperidrose e acne.
Paquidermoperiostose (PDP) ou osteoartropatia hipertrófica primária (OHP) é uma doença hereditária rara descrita pela primeira vez em 1868.1 Sua real incidência é desconhecida,2 mas foram relatados 286 casos em pesquisa no Medline. Predomina em adolescentes do sexo masculino, com uma relação homem:mulher de aproximadamente 7:1.3
A osteoartropatia hipertrófica (OAH) é dividida em formas primárias e secundárias. A PDP é a forma primária, responsável por 3%‐5% de todos os casos de OAH. A OAH secundária, também chamada de OAH pulmonar, está associada a doenças cardiopulmonares subjacentes e malignidades. O diagnóstico de PDP é firmado com base em dados clínicos e radiológicos.1,2
Relata‐se um caso de PDP em paciente jovem do sexo masculino.
Relato do casoPaciente do sexo masculino, 24 anos, relatou espessamento cutâneo na face e no couro cabeludo, desde a adolescência, além de aumento de volume das mãos e dos pés. Referiu também acne desde a adolescência. Negou história familiar de caso semelhante. Ao exame físico identificaram‐se pápulas foliculares eritematosas e pústulas na face, espessamento cutâneo, acentuação dos sulcos faciais com circunvoluções cutâneas (cutis verticis gyrata) na fronte (fig. 1) e couro cabeludo, onde também havia nódulos de consistência amolecida com alopecia sobre suas superfícies, compatível com foliculite abcedante (fig. 2). Mãos e pés com aumento do volume e baqueteamento digital, com unhas em vidro de relógio (fig. 3), além de sudorese aumentada nas mãos e nos pés (hiperidrose). As radiografias de mãos, punhos e joelhos mostraram reação periosteal hipertrófica benigna com alargamento de ossos por hiperostose, além de densificação e aumento da espessura de partes moles adjacentes (fig. 4). Os níveis de fator de crescimento semelhante à insulina‐1 (IGF‐1) apresentavam‐se normais. Com base nas características clínicas (paquidermia, hipocratismo digital e periostose), a forma completa de PDP foi diagnosticada.
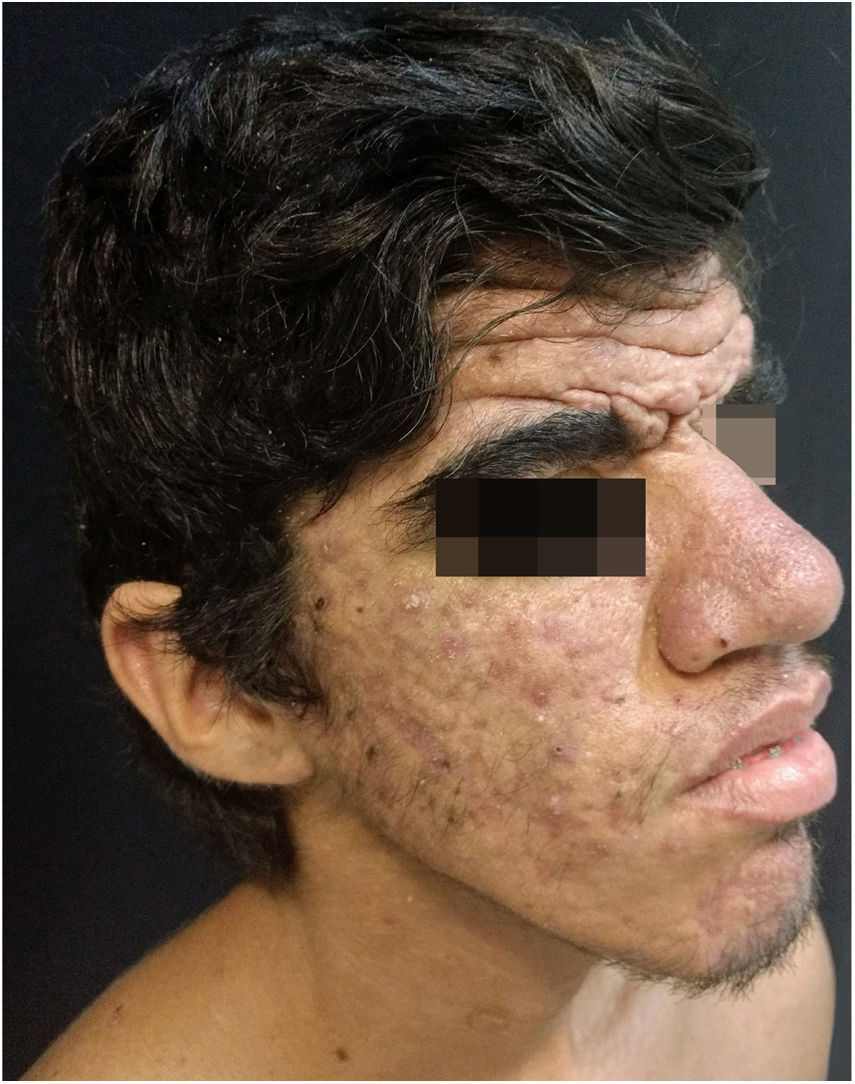
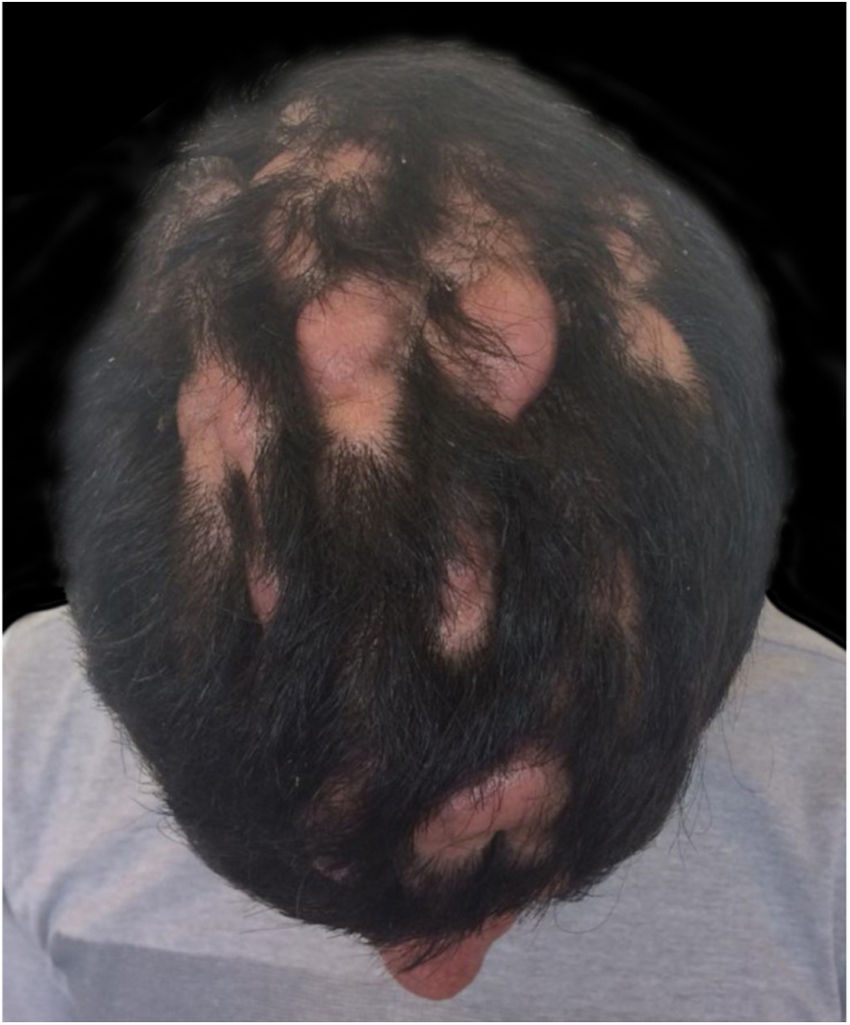
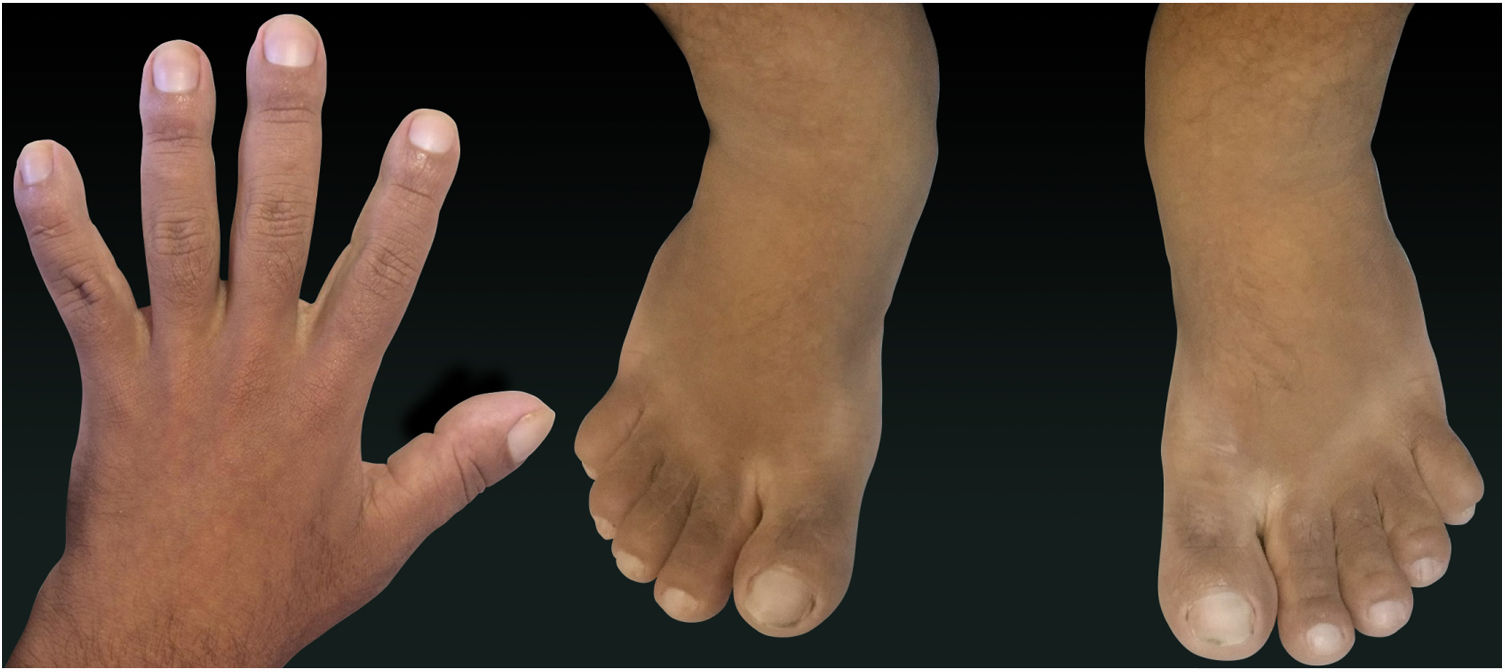
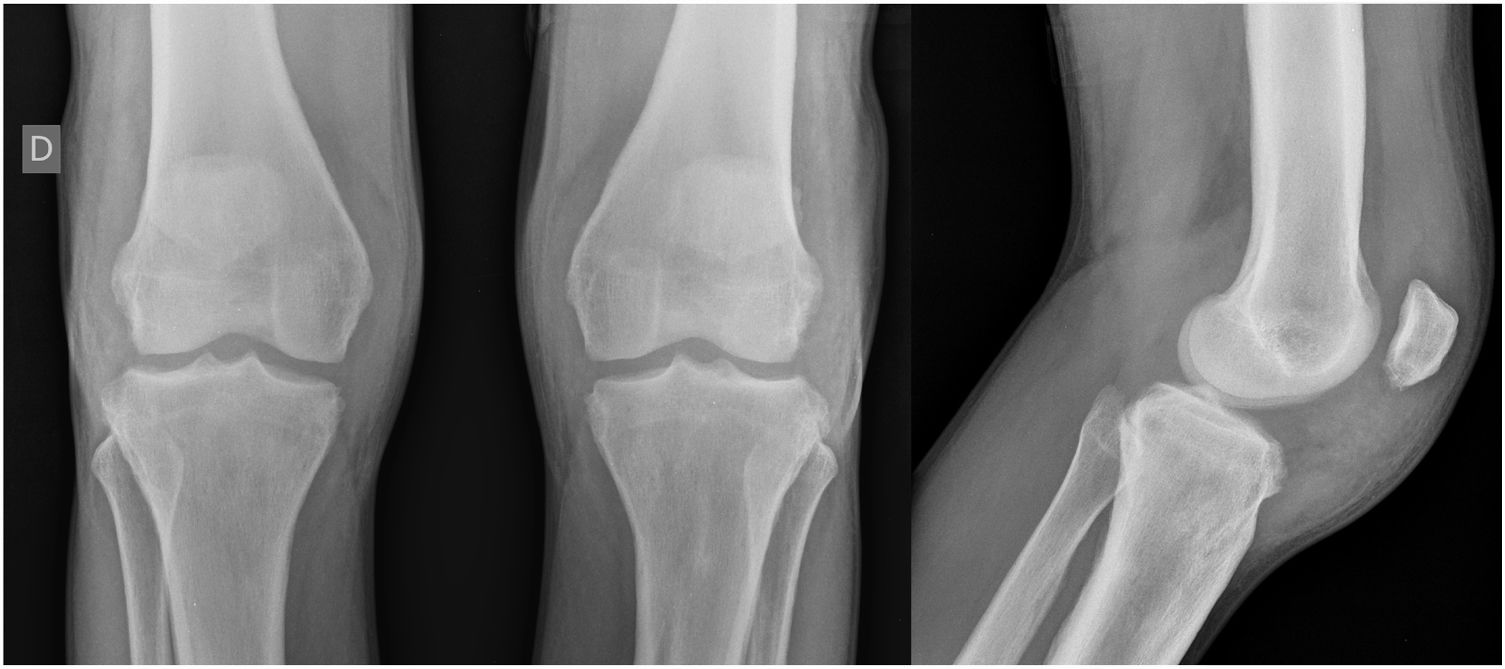
A PDP é uma síndrome genética caracterizada por três sintomas principais: baqueteamento digital, paquidermia (espessamento e enrugamento da pele facial e/ou couro cabeludo), periostose (aumento do tecido periarticular e neoformação óssea subperiosteal). Outras manifestações são: características faciais grosseiras, poliartrite, cutis verticis gyrata (24%), seborreia, ptose palpebral, hiperidrose, acne, artropatia e acro‐osteólise de ossos longos.1,2
As apresentações clínicas geralmente reconhecidas são a forma completa, que envolve os três principais sintomas, a forma incompleta, com periostose sem paquidermia, e a forma frustra, com paquidermia e mínima ou nenhuma anomalia esquelética. Ambas as formas são autossômicas dominantes com penetrância incompleta e herança recessiva tem sido sugerida.1–4 Geralmente o quadro inicia‐se na infância ou adolescência e progride gradualmente por 5‐20 anos, até estabilização.3
A patogênese da PDP ainda não está claramente entendida. A doença foi mapeada para a banda 4q33‐q34 e foram identificadas mutações no gene HPGD, que codifica a 15‐hidroxiprostaglandina desidrogenase, a principal enzima de degradação de prostaglandinas.5,6
Níveis aumentados de prostaglandina E2 (PGE2), resultante de degradação defeituosa devido a mutações nos genes HPGD e SLCO2A1, parecem contribuir para a patogênese da PDP. A gravidade da paquidermia e alterações histológicas associadas foram correlacionadas com níveis séricos de PGE2 e genótipos SLCO2A1. A PGE2 pode mimetizar a atividade de osteoblastos e osteoclastos, que podem ser responsáveis pela acro‐osteólise e formação óssea periosteal. Além disso, os efeitos vasodilatadores locais prolongados da PGE2 podem explicar o baqueteamento digital.7,8
O caso relatado mostra uma forma completa da síndrome em paciente do sexo masculino com quadro iniciado na adolescência (epidemiologia típica), com achados clínicos e radiológicos compatíveis. Apresentava os três achados cardinais, além de outras manifestações associadas, como hiperidrose e acne. Apesar de haver história familiar em um terço dos pacientes com PDP,3 nosso paciente não tinha parentes com características suspeitas desse diagnóstico. Os níveis de IGF‐1 foram normais, contribuíram para descartar hipótese de acromegalia, um importante diagnóstico diferencial.3
Não foi estabelecido um tratamento definitivo para a doença. Os sintomas são manejados com anti‐inflamatórios não esteroidais (AINEs), analgésicos simples e endovenosos, bem como bisfosfonatos. Além disso, o tamoxifeno tem sido relatado como eficaz para artralgia refratária aos AINEs.3
O relato em questão relembra a importância de se considerar PDP como diagnóstico possível na dermatologia e mostra que firmar um diagnóstico de PDP pode ser extremamente desafiador mesmo para médicos mais experientes, sobretudo devido à raridade da doença.
Suporte financeiroNenhum.
Contribuição dos autoresMônica Larissa Padilha Honório: Elaboração e redação do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Guilherme Holanda Bezerra: Elaboração e redação do manuscrito; revisão crítica da literatura.
Vivianne Lira da Câmara Costa: Aprovação da versão final do manuscrito; participação efetiva na orientação da pesquisa; revisão crítica do manuscrito.
Conflitos de interesseNenhum.
