




Fabry disease is a rare lysosomal storage disorder, inherited in an X-linked manner. It is characterized by the deficiency of the enzyme alpha-galactosidase, leading to a buildup of glycosphingolipids in the cells. Angiokeratoma is one of the cutaneous manifestations of this condition, and it helps making the diagnosis. The typical site involves the genital area in men and lumbosacral, buttocks and trunk region in both sexes. We report a case of genital angiokeratoma in a woman with Fabry disease. The diagnosis is through molecular analysis and, when made early, starting treatment reduces the morbidity and mortality of the disease. Thus, the dermatologist has an important role in the identification of angiokeratoma as a cutaneous marker, and the knowledge of its different presentations is essential for the early diagnosis and management of Fabry disease.
Fabry disease (FD) is a rare storage disorder, first described in 1898 as angiokeratoma corporis diffusum by Anderson and Fabry.1,2 Subsequently, it was described as a systemic metabolic disease, affecting cells of the vascular endothelium, smooth muscle, myocardium, renal epithelium and central nervous system. The time between the onset of symptoms and the diagnosis can be longer than 10 years.3
FD is a lysosomal disease, inherited in an X-linked manner.4 The affected gene is called GLA and leads to the deficiency of the enzyme alpha galactosidase, with subsequent buildup of glycosphingolipids in the vascular endothelium, smooth muscle, myocardium, renal epithelium and central nervous system. In male patients, the gene has a high penetrance, originating the so called classic phenotype of the disease.5 The diagnosis in this population is through the dosage of the enzymatic activity of alpha galactosidase.6 In female patients, the disease has variable expression due to the random inactivation of one of the X chromosomes, according to the hypothesis of Lyon.7 Currently, heterozygous patients are not considered only carriers, since they present systemic changes related to the disease and increased mortality. In women, the diagnosis is through molecular analysis of the GLA gene, since the dosage of the enzyme can be abnormal, as in men, or normal, as in healthy individuals.7
The diagnosis should be suspected when finding the following symptoms and signs: acroparesthesia (burning pain in the hands and feet), hearing loss, cornea verticillata, recurrent headaches, abdominal pain, chronic diarrhea, progressive renal failure, cardiomyopathy, psychiatric conditions, ischemic encephalopathy and angiokeratomas. Angiokeratomas (AK) can present in different ways and help in the identification of patients affected by FD.8 The disease can become severe and lethal if there is brain, renal or cardiac involvement. Early diagnosis and enzyme replacement therapy (ERT) reduce morbidity and mortality in these patients.5
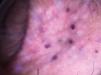
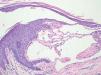
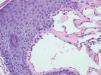
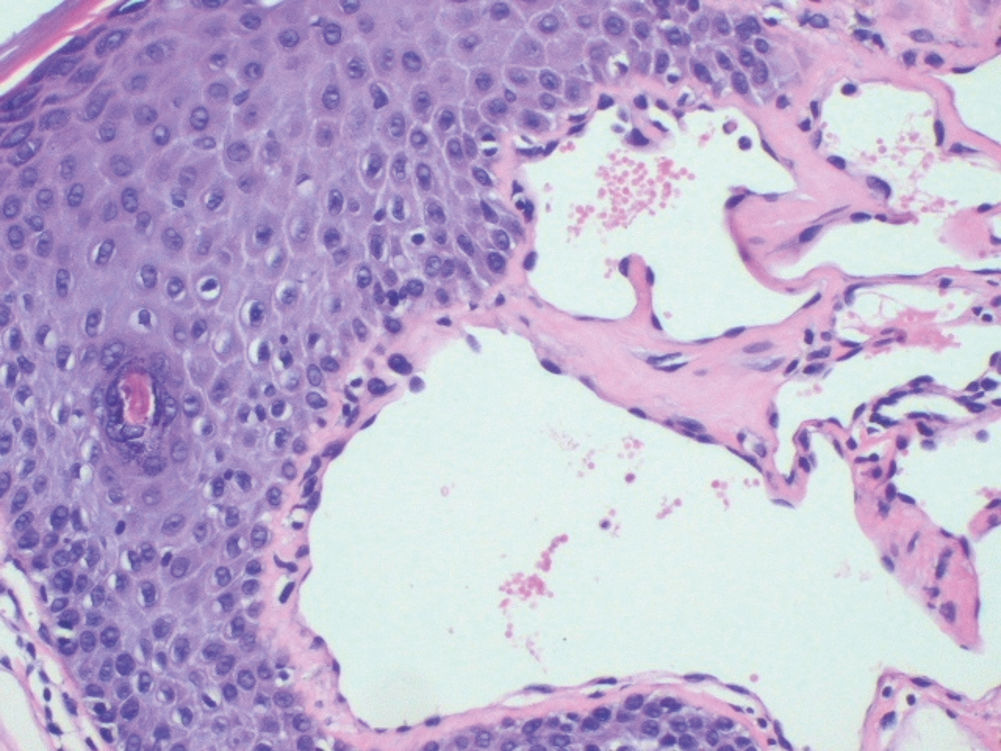
Case ReportFemale, 52-year-old patient with FD, diagnosed two years prior to the consultation through family screening. She was the first daughter of a non-blood-related relationship, and her brother was the index case of the family. He had had renal transplantation and had fixed lymphedema in the lower limbs, AK on the back and depression. The patient’s son, who was also diagnosed with FD during screening, had acroparesthesia and hypohidrosis with intolerance to exercise. The patient reported daily headaches since childhood and pain in the extremities since she was a teenager. On examination, she had erythematous-purple papules, with a slightly keratotic surface on the vulva (Figure 1). Dermoscopy revealed multiple blue-red lakes (Figure 2). One of the papules was biopsied, revealing vascular dilation in the papillary dermis, associated to overlying acanthosis (Figures 3 and 4). Both tests supported the diagnosis of AK. The family is undergoing ERT, with a good clinical response.



AKs are an important cutaneous marker in the screening of FD patients and can present in many different forms. They are present in 66% of affected men and 36% of women, who can have early and atypical manifestations of the disease. They are papules varying in or from red to black, with 1mm to 5mm diameter, with a smooth1 or keratotic surface. They usually appear in adolescence, and progressively increase. The affected sites are the genital region in men and lumbosacral, buttocks and trunk region in both sexes. Subsequently, they can appear on the lips, navel, nails and palms. Men have a larger number of lesions. Women rarely present AKs on the trunk and genital regions.2 In a Medline and Embase database review, we did not find any similar cases in the literature till present.
Among the dermatologic lesions we can find — besides AKs —telangiectasias, lymphedema and anhidrosis. The patient had angiomas. The son had hypohidrosis, and the brother had fixed lymphedema of the lower limbs.
The dermoscopy is helpful in detecting multiple AKs that are not always seen on clinical examination alone. In this case, they had a multilobular vascular configuration, described as a well-defined red lake, with white-yellow areas corresponding to hyperkeratosis.3 According to the description in the literature, some dermoscopic findings suggest possible associated lesions, such as darkened lakes associated to thrombosis, hemorrhagic crusts, bleeding and, finally, erythematous areas and local inflammation.
Histopathology reveals dilated blood vessels in the papillary dermis with overlying epidermal hyperplasia, confirming the clinical and dermoscopic findings.4
The differential diagnoses for AK include angioma, pyogenic granuloma, common warts, Spitz nevus, seborrheic keratosis and melanoma.5,6 They should also be differentiated from other types of AK [acral (Mibelli), genital (Fordyce), circumscribed neviform, solitary and corporis diffusum]. A positive family history for FD (brother and sons), associated to symptoms and signs found, lead to the molecular analysis, that confirmed the diagnosis and ruled out the above-mentioned differentials.
ERT is available since 2001 for FD and is the only therapeutic option till present.7–9 In Brazil, it is indicated to patients older than 18 years of age with proteinuria, myocardial hypertrophy, cerebral ischemia, gastrointestinal symptoms and acroparesthesia.8,10 It improves the overall prognosis and can also stabilize and reduce the cutaneous lesions. If the AK is an aesthetic concern, treatment with electrocoagulation and ablative laser is indicated.
Our patient is under treatment with ERT for 12 months and improved from the acroparesthesia. For the time being, the AK is being monitored clinically.
In conclusion, we can say that AK is an important cutaneous marker in screening patients for FD, and the knowledge of this lesions is helpful in identifying the disease early.7,10
Aknowledgements:We thank Dr. Alexandre Ozores Michalany, who performed the histopathology examination and made available the images in this article.
Financial support: None.
Conflict of interest: None.
