





Uma menina chinesa de 15 anos apresentou história de 10 anos de máculas marrom‐claras assintomáticas e unilaterais que afetavam o braço direito e o lado direito do tronco. Não havia histórico de lesões cutâneas ou inflamação. O histórico médico e familiar não foi significativo. No exame físico, foram observadas máculas atróficas lineares hiperpigmentadas no braço e tronco direitos que seguiam as linhas de Blaschko e acometiam os aspectos anterior e posterior. A pele estava levemente atrófica à palpação. Não foram observados sinais de enrijecimento ou inflamação (fig. 1A‐B). Os exames laboratoriais – inclusive hemograma, taxa de sedimentação de eritrócitos, teste da função hepática, perfil renal e anticorpos antinucleares – foram todos negativos ou dentro da faixa normal. A biópsia de lesão evidenciou epiderme normal com pigmentação aumentada na camada basal, colágeno dérmico mais compacto e infiltração linfocitária perivascular de pequeno grau na derme superior (fig. 2). A dermatoscopia evidenciou rede marrom‐clara com margens pouco demarcadas. Foi estabelecido o diagnóstico de atrofodermia linear de Moulin (ALM) e iniciou‐se o tratamento tópico com creme de halometasona a 0,5% e creme de hidroquinona a 2% por dois meses, sem melhora.
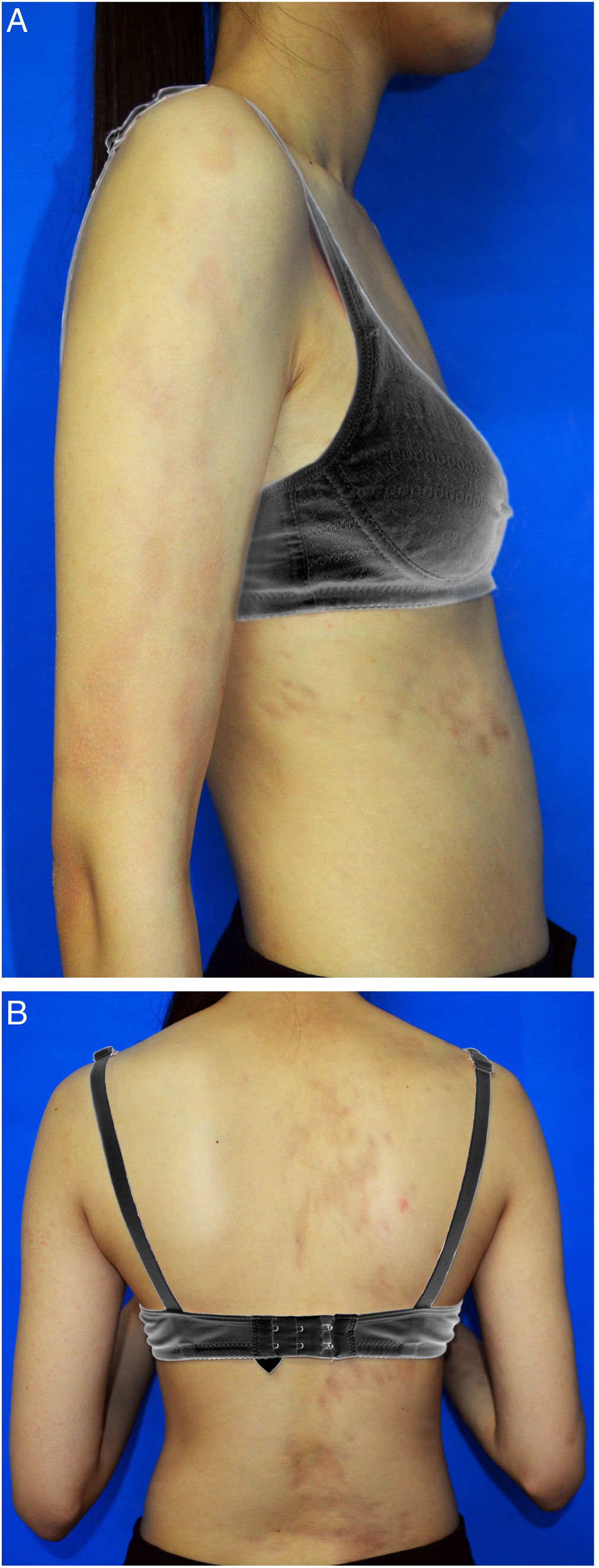
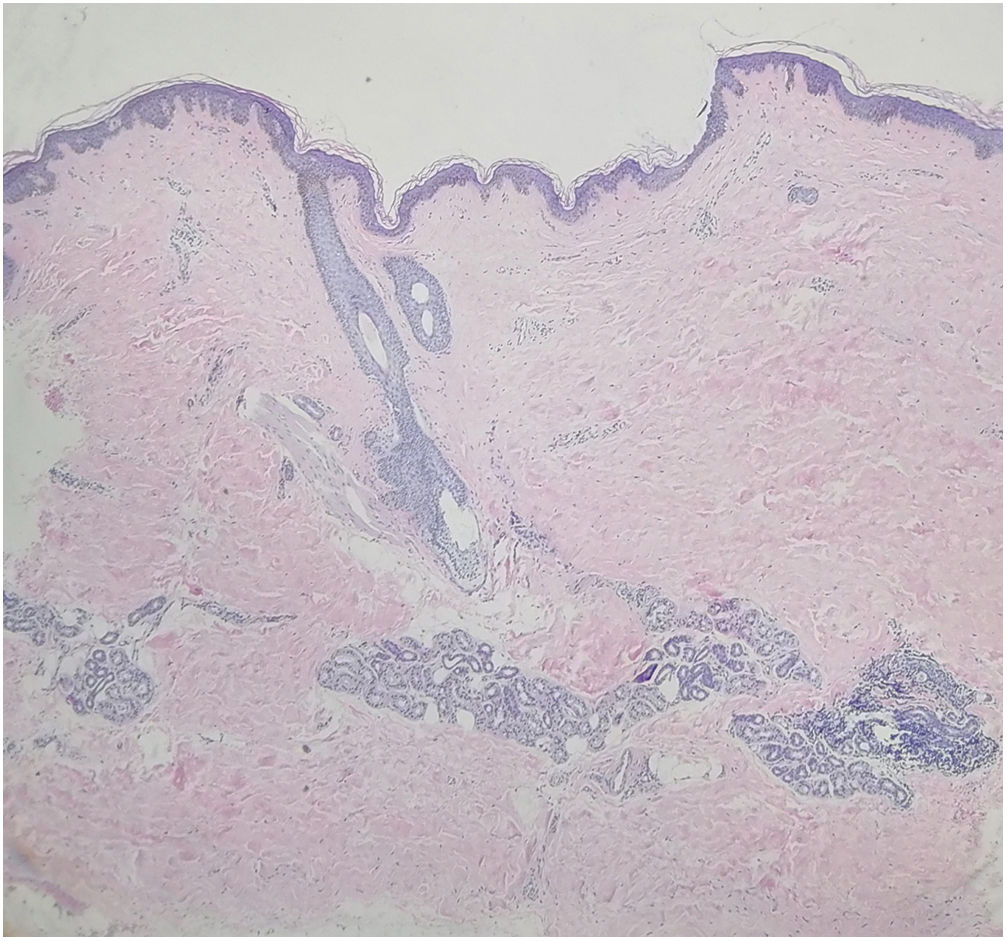
A ALM é entidade clínica rara e distinta, caracterizada por lesões cutâneas unilaterais, hiperpigmentadas e atróficas em faixa, que seguem as linhas de Blaschko, sem inflamação prévia ou aparência esclerótica. A doença recebe o nome do pesquisador que, em 1992, relatou o caso de cinco pacientes com faixas pigmentadas mais ou menos atróficas ao longo das linhas de Blaschko.1 A ALM geralmente evolui como lesão atrófica linear nos primeiros meses, deixa então de progredir e persiste. Sua etiologia permanece incerta. Todos os casos relatados até o momento foram esporádicos. Ela pode estar ligada a um mosaicismo genético ou autoimunidade. Um estudo do componente atrófico da ALM por ultrassonografia revelou que a redução do volume subcutâneo, não a diminuição da derme, foi a causa da aparência atrófica.2 Embora sua manifestação clínica seja singular, a histopatologia da ALM é bastante discreta. A coloração com hematoxilina & eosina geralmente evidencia hiperpigmentação apenas na camada basal da epiderme, ausência de colágeno anormal ou fibras elásticas alteradas na derme ou de qualquer inflamação óbvia.1 Pode haver certo grau de infiltrado linfocitário perivascular, acantose, adelgaçamento da epiderme, alterações no colágeno dérmico e redução ou fragmentação do tecido elástico.2
López et al.3 propuseram os seguintes critérios diagnósticos para a ALM: 1) Início durante a infância ou adolescência; 2) Desenvolvimento de lesões unilaterais hiperpigmentadas, levemente atróficas, que seguem as linhas de Blaschko no tronco ou nos membros; 3) Ausência de inflamação prévia ou de esclerodermia subsequente; 4) Curso clínico estável, não progressivo, sem padrão de remissão; 5) Achados histológicos de hiperpigmentação da camada basal da epiderme e derme normal com tecido conjuntivo e fibras elásticas inalterados. Até o momento, mais de 30 casos de ALM foram relatados na literatura. No entanto, esse diagnóstico pode ter sido superestimado. Se os critérios diagnósticos forem rigorosamente seguidos, alguns casos não podem ser classificados como ALM, pois esses autores relataram achados histológicos compatíveis com outras entidades clínicas.3
A ALM deve ser diferenciada da atrofodermia de Pasini e Pierini (APP), que apresenta configuração, atrofia e hiperpigmentação semelhantes, mas não segue as linhas de Blaschko. Além disso, a ALM é diferente da morfeia linear, que geralmente apresenta inflamação, enrijecimento ou esclerodermia anteriores.
Histopatologicamente, a morfeia apresenta feixes de colágeno compactados e orientados horizontalmente, com perda progressiva dos anexos cutâneos e da gordura subcutânea. No entanto, ainda não existe um consenso se a ALM é uma entidade distinta. Existem muitas semelhanças clínicas e histológicas entre ALM, APP e morfeia; portanto, alguns autores sugerem que essas doenças são parte de um espectro e que a ALM pode não ser uma entidade distinta.4 A ALM pode ser uma variante Blaschko‐linear da APP, que por sua vez pode ser considerada uma forma abortiva de morfeia.4
Não existe tratamento eficaz para a ALM. Corticosteroides tópicos e heparina não foram bem‐sucedidos. Alguns tratamentos experimentais levaram a uma resposta parcial: calcipotriol tópico, metotrexato sistêmico ou aminobenzoato e terapia intralesional com plasma rico em plaquetas.5 O presente relato apresenta um caso de ALM com características clínicas e histopatológicas clássicas.
Suporte financeiroNenhum.
Contribuição dos autoresLi‐wen Zhang: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; revisão crítica da literatura; revisão crítica do manuscrito.
Meng‐sha Ma: Obtenção, análise e interpretação dos dados; revisão crítica do manuscrito.
Tao Chen: Aprovação da versão final do manuscrito; concepção e planejamento do estudo.
Li‐xin Fu: Revisão crítica da literatura.
Conflitos de interesseNenhum.
