

Amiloidose cutânea discrômica (ACD, OMIM #617920) é doença autossômica recessiva ou dominante rara causada pela mutação do gene da glicoproteína não metastática B (GPNMB) e caracterizada por hiper/hipopigmentação mosqueada clinicamente difusa e deposição dérmica de amiloide. Até o momento, 16 mutações diferentes de GPNMB de 28 linhagens de ACD foram documentadas na literatura em inglês.1–5 O presente caso descreve uma família chinesa com ACD autossômica recessiva com mutações heterozigóticas compostas de GPNMB.
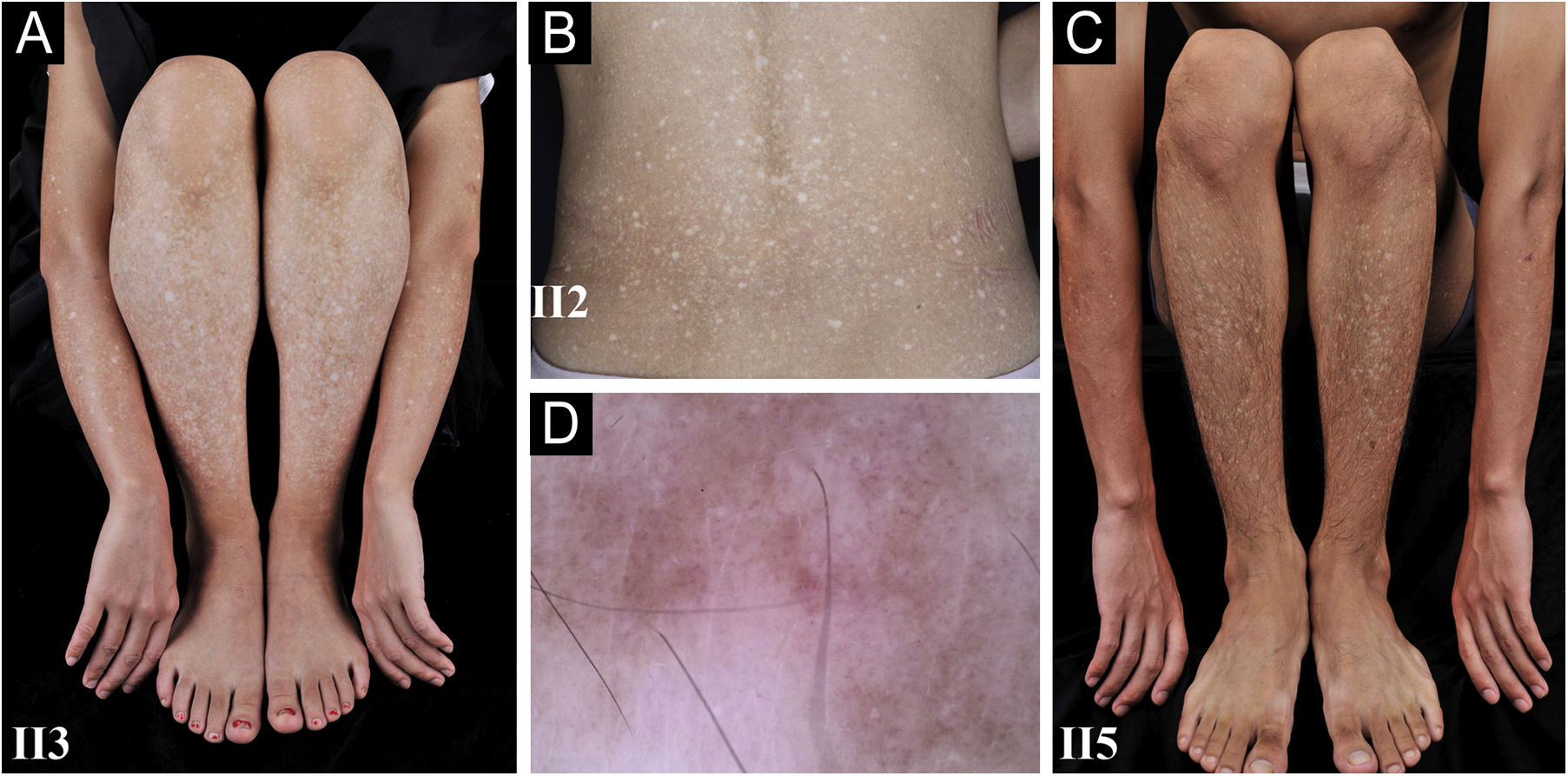
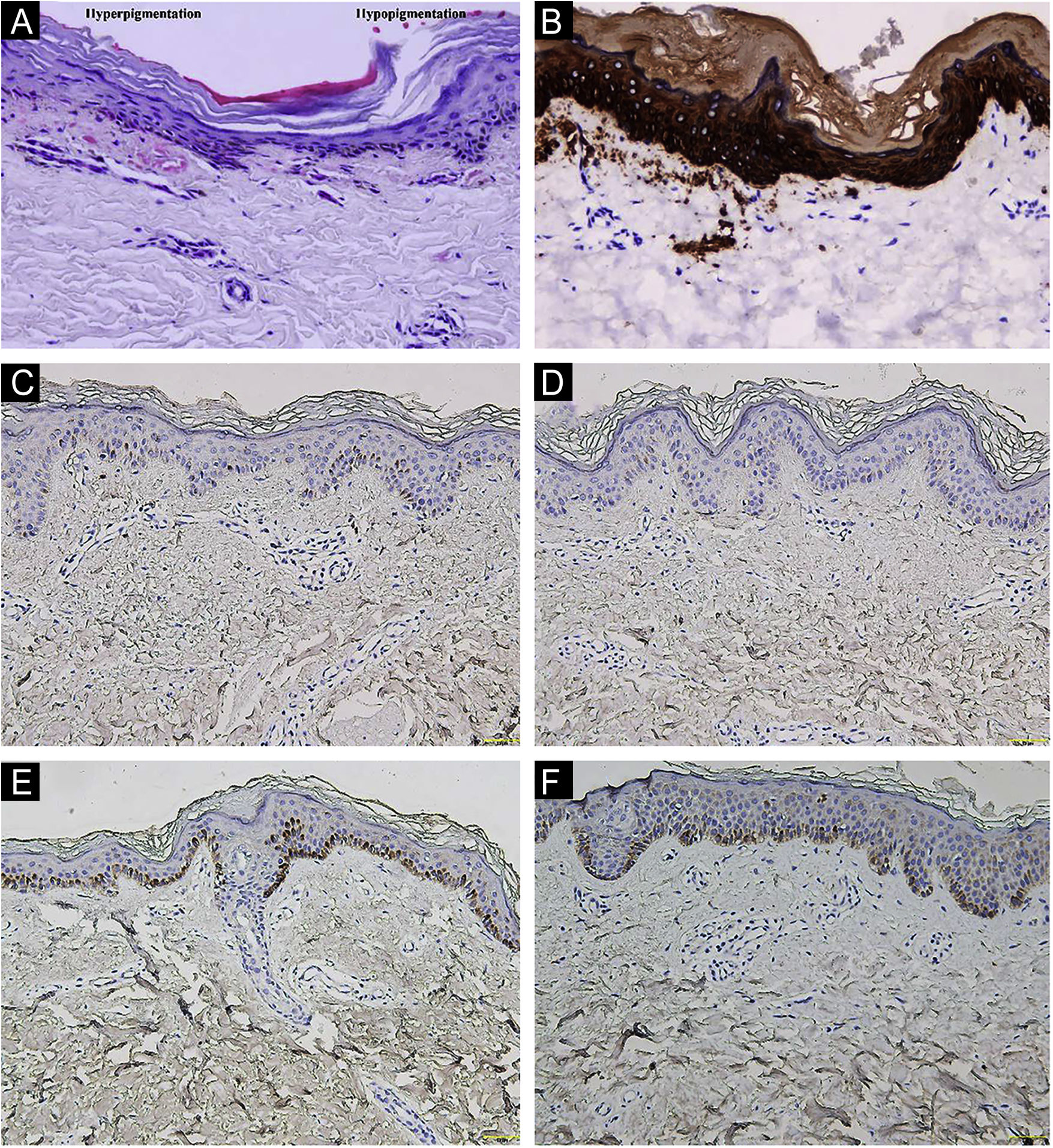
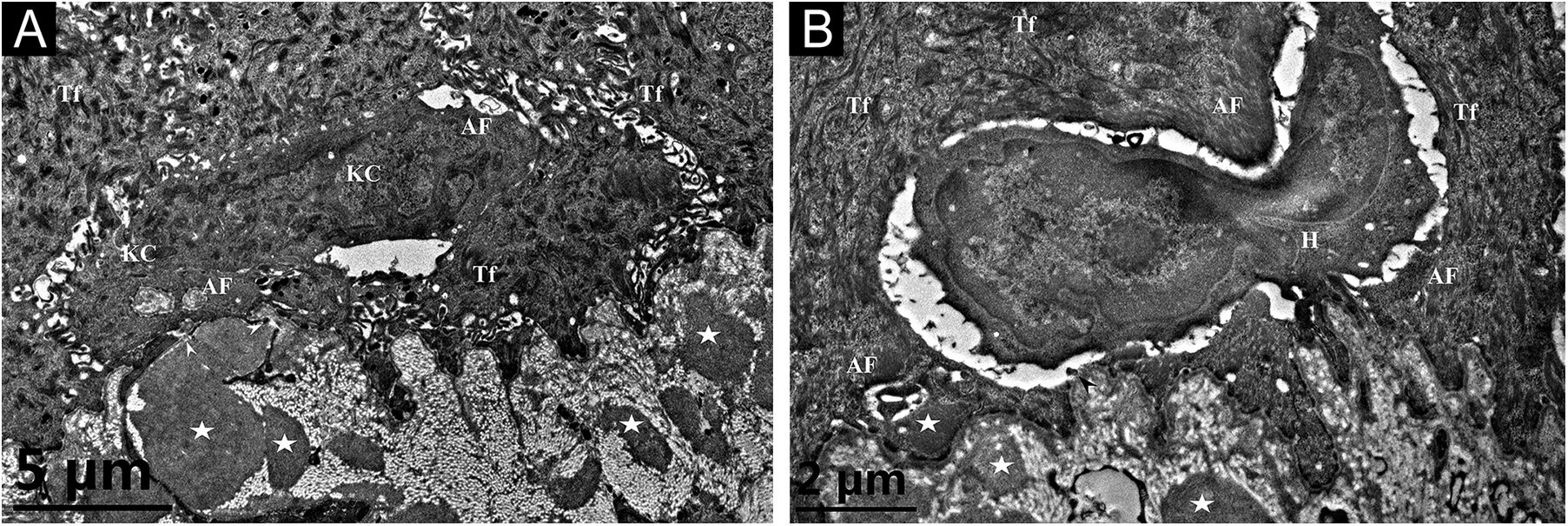
Paciente feminina, chinesa, de 26 anos, apresentou história de 21 anos de pigmentação mosqueada generalizada assintomática. A despigmentação iniciou‐se nos membros e espalhou‐se progressivamente por todo o corpo. Várias bolhas pruriginosas ocorreram de forma recorrente nos braços durante o verão e cicatrizaram espontaneamente sem deixar cicatrizes, mas fotossensibilidade estava ausente. O exame dermatológico mostrou numerosas máculas reticuladas hiper/hipopigmentadas afetando quase todo o corpo, com leve acometimento da face e região cervical, poupando o dorso, mãos e pés (fig. 1A). Cabelo, unhas, dentes e mucosas eram normais. Dois irmãos apresentavam lesões semelhantes (fig. 1B‐C), mas outros membros da família, incluindo pais não consanguíneos, não foram afetados. A dermatoscopia mostrava máculas brancas, irregulares, mal definidas, circundadas por pigmentação acastanhada e vasos lineares indistintos (fig. 1D). Os exames laboratoriais, incluindo hemograma completo, urinálise, perfil bioquímico e de anticorpos antinucleares, radiografia de tórax e ultrassonografia abdominal não mostraram resultados dignos de nota. A microscopia óptica de lesão no braço revelou epiderme hiperceratótica e parcialmente atrófica com hipopigmentação da camada basal na área hipopigmentada, e epiderme hiperceratótica com pequena hiperpigmentação e degeneração vacuolar focal da camada basal, depósitos eosinofílicos amorfos e melanófagos esparsos estavam presentes na derme papilar na área hiperpigmentada (fig. 2A). A coloração pelo método de Masson‐Fontana mostrou hipermelanose na área hiperpigmentada. Os depósitos dérmicos foram melhor evidenciados na coloração pelo método do vermelho do Congo e na imunomarcação com citoqueratina CK 34βE12 de alto peso molecular (fig. 2B). Biópsias de pele adicionais foram obtidas de outros membros da família (II2, II4 e II5). A coloração pelo método do vermelho do Congo e a imunomarcação com CK 5/6 exibiram depósitos amiloides abundantes na lesão hiperpigmentada e em pequena quantidade na lesão hipopigmentada em três indivíduos afetados, e ausentes em um portador, II4, com a mutação c.565C>T. Na imuno‐histoquímica, a expressão citoplasmática de GPNMB nas camadas basal e suprabasal foi fraca na lesão hiperpigmentada e ausente na lesão hipopigmentada de pacientes com ACD, e moderada na pele do antebraço do portador II4 com a mutação c.565C>T e no controle normal (fig. 2C‐F). A microscopia eletrônica revelou agregados fibrilares intracitoplasmáticos em queratinócitos basais degenerados e destruição parcial da lâmina basal e da citomembrana entre os depósitos amiloides superiores e as células basais em algumas áreas. Corpos fibrilares homogêneos eram circundados por feixes de colágeno e processos fibroblásticos e histiocíticos na derme papilar (fig. 3).
 Hiperpigmentação difusa mesclada com numerosas máculas hipopigmentadas nos membros e região dorsal em três pacientes com ACD, sem comprometimento do dorso das mãos e pés. (D) A dermatoscopia apresentava máculas brancas, irregulares, mal definidas, circundadas por pigmentação acastanhada e vasos lineares indistintos (ampliação original 60×).")
Observação clínica e dermatoscópica. (A‐C) Hiperpigmentação difusa mesclada com numerosas máculas hipopigmentadas nos membros e região dorsal em três pacientes com ACD, sem comprometimento do dorso das mãos e pés. (D) A dermatoscopia apresentava máculas brancas, irregulares, mal definidas, circundadas por pigmentação acastanhada e vasos lineares indistintos (ampliação original 60×).
 A coloração pela Hematoxilina & eosina mostrou epiderme hiperceratótica com pequena hiperpigmentação e degeneração vacuolar focal da camada basal, e depósitos eosinofílicos amorfos e melanófagos esparsos na derme papilar (lado esquerdo); e epiderme hiperceratótica e parcialmente atrófica com hipopigmentação da camada basal (lado direito; ampliação original 200×). (B) A imunomarcação com citoqueratina 34βE12 revelou depósitos amiloides na derme superior (ampliação original 200×). (C‐F) A imunorreatividade de GPNMB foi fraca na lesão hiperpigmentada (C) e ausente na lesão hipopigmentada (D) do paciente II5, e moderada na pele do antebraço do portador II4 com mutação c.565C>T (E) e no controle normal (F; ampliação original 200×).")
Estudo histopatológico e imuno‐histoquímico. (A) A coloração pela Hematoxilina & eosina mostrou epiderme hiperceratótica com pequena hiperpigmentação e degeneração vacuolar focal da camada basal, e depósitos eosinofílicos amorfos e melanófagos esparsos na derme papilar (lado esquerdo); e epiderme hiperceratótica e parcialmente atrófica com hipopigmentação da camada basal (lado direito; ampliação original 200×). (B) A imunomarcação com citoqueratina 34βE12 revelou depósitos amiloides na derme superior (ampliação original 200×). (C‐F) A imunorreatividade de GPNMB foi fraca na lesão hiperpigmentada (C) e ausente na lesão hipopigmentada (D) do paciente II5, e moderada na pele do antebraço do portador II4 com mutação c.565C>T (E) e no controle normal (F; ampliação original 200×).
 Filamentos citoplasmáticos semelhantes a substância amiloide (AF) em dois queratinócitos basais degenerados (KC), tonofilamentos (Tf) em queratinócitos normais adjacentes, ruptura parcial (pontas de setas) da lâmina basal e citomembrana entre o depósito amiloide superior e a célula basal degenerada, e corpos fibrilares (asteriscos) na derme papilar (ampliação original 8.000×). (B) Um histiócito (H) está localizado entre dois queratinócitos basais parcialmente degenerados contendo AF e Tf; observa‐se ruptura parcial (ponta de seta) da lâmina basal e da citomembrana, e corpos fibrilares (asteriscos) na derme papilar (ampliação original 15.000×).")
Microscopia eletrônica de transmissão. (A) Filamentos citoplasmáticos semelhantes a substância amiloide (AF) em dois queratinócitos basais degenerados (KC), tonofilamentos (Tf) em queratinócitos normais adjacentes, ruptura parcial (pontas de setas) da lâmina basal e citomembrana entre o depósito amiloide superior e a célula basal degenerada, e corpos fibrilares (asteriscos) na derme papilar (ampliação original 8.000×). (B) Um histiócito (H) está localizado entre dois queratinócitos basais parcialmente degenerados contendo AF e Tf; observa‐se ruptura parcial (ponta de seta) da lâmina basal e da citomembrana, e corpos fibrilares (asteriscos) na derme papilar (ampliação original 15.000×).
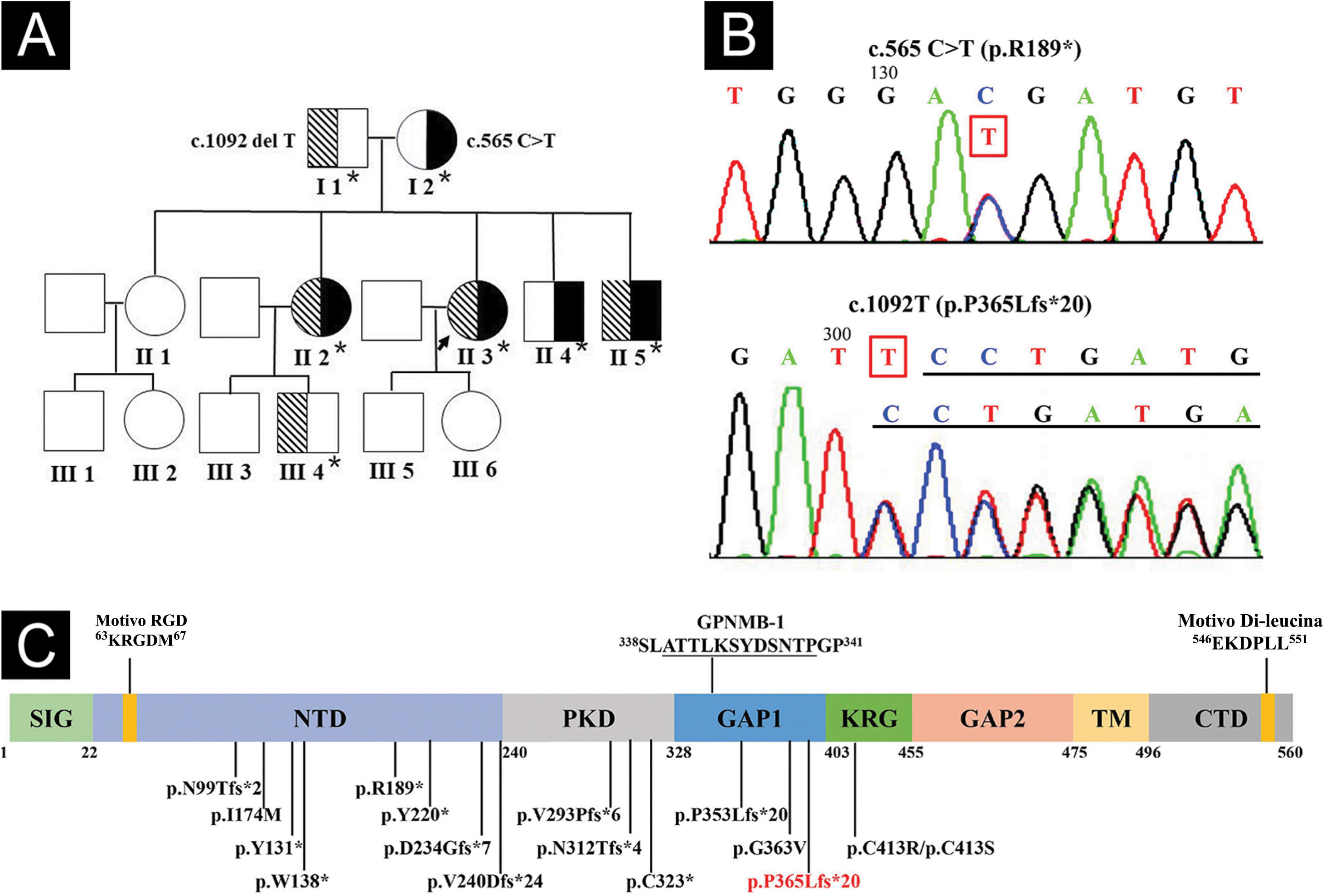
O sequenciamento do exoma completo e o de Sanger do DNA do sangue periférico identificaram mutações heterozigóticas compostas de c.565C> T (p.R189 *) no éxon 5 e c.1092delT (p.P365Lfs * 21) no éxon 7 de GPNMB em três irmãos afetados (fig. 4A‐B), que eram respectivamente derivadas da mãe e do pai. Três irmãos afetados foram diagnosticados com ACD, e as lesões cutâneas do probando permaneceram estáveis após 4,5 anos de seguimento.
 Linhagem do probando. Os asteriscos representam os participantes com testes genéticos. (B) O sequenciamento de Sanger do probando mostrou mutações heterozigóticas compostas de c.565C>T no éxon 5 e c.1092delT no éxon 7 de GPNMB. (C) Estrutura e 16 mutações da proteína GPNMB humana originadas de 17 GPNMB mutantes (p.V240Dfs*24 derivados dos mutantes c.717_718delTG e c.719_720delTG). Uma nova mutação c.1092delT nessa linhagem está marcada em vermelho. Mutantes GPNMB‐1 (uma isoforma de splice de GPNMB) com inserção in frame de 12 aminoácidos (sublinhada) e motivos tipo RGD e Di‐leucina também são mostrados. SIG, domínio de sequência de sinais; NTD, domínio N‐terminal; PKD, domínio de doença renal policística‐símile; KRG, domínio Kringle‐ símile; TM, domínio transmembrana; CTD, domínio citoplasmático C‐terminal.")
Genograma e sequenciamento de GPNMB e estrutura proteica. (A) Linhagem do probando. Os asteriscos representam os participantes com testes genéticos. (B) O sequenciamento de Sanger do probando mostrou mutações heterozigóticas compostas de c.565C>T no éxon 5 e c.1092delT no éxon 7 de GPNMB. (C) Estrutura e 16 mutações da proteína GPNMB humana originadas de 17 GPNMB mutantes (p.V240Dfs*24 derivados dos mutantes c.717_718delTG e c.719_720delTG). Uma nova mutação c.1092delT nessa linhagem está marcada em vermelho. Mutantes GPNMB‐1 (uma isoforma de splice de GPNMB) com inserção in frame de 12 aminoácidos (sublinhada) e motivos tipo RGD e Di‐leucina também são mostrados. SIG, domínio de sequência de sinais; NTD, domínio N‐terminal; PKD, domínio de doença renal policística‐símile; KRG, domínio Kringle‐ símile; TM, domínio transmembrana; CTD, domínio citoplasmático C‐terminal.
Mutações heterozigóticas compostas de GPNMB na mutação c.565C> T conhecida e uma nova mutação c.1092delT foram encontradas nesta família. Assim, 17 mutações de GPNMB associadas a ACD de 29 linhagens de ACD (incluindo o presente caso) foram identificadas, a saber: sete (41,2%) frameshift, cinco (29,4%) nonsense, quatro (23,5%) missense e uma (5,9%) mutação splice (fig. 4C).1–5 O mutante c.565C>T é mutação nonsense comum na população do Leste Asiático e mutação fundadora em pacientes chineses com ACD, resultando em terminação prematura (p.R189*) no domínio N‐terminal de GPNMB.1,2 Além disso, semelhante ao mutante c.1056delT (p.P353Lfs*20),1 c.1092delT(p.P365Lfs*21) pode ser uma nova mutação frameshift causando terminação prematura entre domínios da doença renal policística‐símile e Kringle‐ símile de GPNMB.
GPNMB é altamente expresso em melanócitos e é fundamental para a formação de melanossomas.1 A expressão de GPNMB estava diminuída em lesões hipopigmentadas de pacientes com ACD e vitiligo.2 Os portadores heterozigotos de GPNMB com c.700+5G>T apresentaram hiperpigmentação leve e expressão negativa de GPNMB e nenhuma deposição de amiloide em uma linhagem semidominante.3 Entretanto, um portador com a mutação c.565C>T manifestou fenótipo normal e expressão de GPNMB na presente linhagem com ACD recessiva. Esses resultados sugerem que a haploinsuficiência do GPNMB não pode contribuir para o fenótipo da ACD.2
Os depósitos amiloides eram abundantes na lesão hiperpigmentada e escassos na lesão hipopigmentada e imunorreativos com CK5/6 e CK34βE12.1,4 A microscopia eletrônica revelou corpos fibrilares homogêneos na derme papilar e agregados fibrilares intracitoplasmáticos em queratinócitos degenerados.1,3 Meios condicionados de melanócitos silenciados para GPNMB aumentaram a apoptose de queratinócitos.3 Esses resultados indicam que queratinócitos degenerados e necróticos podem contribuir para a formação de amiloide.1 Embora as mutações associadas à ACD possam causar localização aberrante de GPNMB,2 seu papel na patogenia da ACD ainda precisa ser elucidado.
Suporte financeiroEste estudo recebeu suporte financeiro do Discipline Construction Project of Guangdong Medical University (4SG21277P).
Contribuição dos autoresCi‐Juan Zhong: Concepção e planejamento do estudo; obtenção, análise e interpretação dos dados; elaboração e redação do manuscrito.
Fang‐Gu Li: Concepção e planejamento do estudo; obtenção, análise e interpretação dos dados; elaboração e redação do manuscrito.
Wen Li: Obtenção, análise e interpretação dos dados.
Yi‐Ming Fan: Concepção e planejamento do estudo; obtenção, análise e interpretação dos dados; revisão crítica do manuscrito; aprovação da versão final do manuscrito.
Como citar este artigo: Zhong CJ, Li FG, Li W, Fan YM. Amyloidosis cutis dyschromica caused by compound heterozygous GPNMB mutations in a Chinese pedigree. An Bras Dermatol. 2025;100. https://doi.org/10.1016/j.abd.2024.01.009.
Trabalho realizado no Guangdong Medical University, Dongguang, Guangdong, China.
