








A lipoidoproteinose é doença autossômica recessiva rara, caracterizada por depósitos hialinos de material PAS+ nos tecidos, em decorrência de mutações no gene ECM1. Examinamos ultraestruturalmente a pele de uma criança de 6 anos afetada com essa enfermidade. Na microscopia óptica, foram identificados depósitos hialinos PAS+, os quais se apresentavam mais acentuados na derme papilar. Na microscopia eletrônica de varredura da derme, demonstrou‐se acentuada compactação da derme papilar e depósitos fibrilares na derme média. Na microscopia eletrônica de transmissão fica evidente o depósito dérmico de material fibrilar, formando agrupamentos, aderidos a fibras elásticas, entre os feixes colágenos e as fibras colágenas, e também preenchendo o citoplasma dos fibroblastos dérmicos.
A lipoidoproteinose (OMIM 247100), também chamada de doença de Urbach‐Wiethe, ou hyalinosis cutis et mucosae, é uma genodermatose rara, de caráter autossômico recessivo, com expressão variada. Pode comprometer múltiplos sistemas, como mucosas e órgãos internos e apresenta importante acometimento cutâneo.1
Sua ocorrência se deve a mutações do gene extracellular matrix protein 1 (ECM1).2,3 O acometimento cutâneo é caracterizado por depósito de material hialino na derme papilar, com reação positiva ao ácido periódico de Schiff (PAS).
Em seu quadro clínico clássico, de curso crônico e benigno, os pacientes exibem sinais e sintomas desde a infância, com rouquidão e dificuldade na fonação (por acometimento da laringe), seguida de fragilidade cutânea e lesões varioliformes. Há também acometimento neurológico, pela presença de calcificações no sistema nervoso central. O envolvimento das amígdalas é considerado um sinal patognomônico. Apesar do curso benigno na maioria dos indivíduos, a lipoidoproteinose não conta com tratamento definitivo, que é realizado por abordagens sintomáticas nos pacientes acometidos.
Examinamos um menino de 6 anos de idade, caucasiano, acompanhado desde o pré‐natal por mielomeningocele, encaminhado ao departamento de Dermatologia em função de achados compatíveis com lipoidoproteinose em uma tomografia craniana de controle.
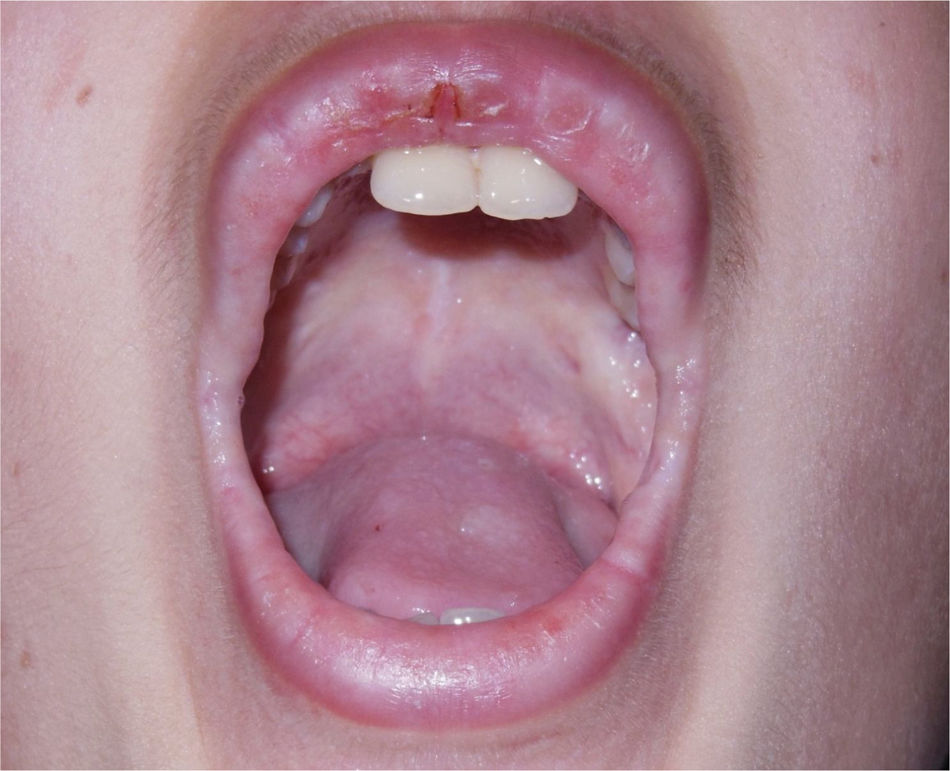
O paciente apresentou choro baixo desde o nascimento e rouquidão ao aprender a falar, com piora gradativa nos últimos anos. Ao exame dermatológico, notava‐se espessamento cutâneo generalizado e aspecto foto envelhecido na face. Foram identificadas placas micropapulosas xantomatosas de brilho céreo na fronte, nas faces laterais do pescoço, no dorso, nos cotovelos e nos joelhos. Na fronte, apresentava cicatrizes acneiformes atróficas confluentes. Nas bordas palpebrais era possível observar numerosas pápulas diminutas enfileiradas ao longo de suas margens sem rarefação ciliar – a chamada blefarite moniliforme. Na cavidade oral, encontramos língua de superfície irregularmente despapilada, frênulo curto e placas brilhantes ora esbranquiçadas, por vezes xantomatosas, nas porções centrais do palato (fig. 1), na língua e no assoalho da boca.
Foi realizada biópsia cutânea por punch da região cervical, a qual foi processada para microscopia óptica (MO), com coloração por Hematoxilina & eosina e PAS. Um pequeno fragmento foi desidratado, para que a derme fosse examinada com microscopia eletrônica de varredura (MEV); outro fragmento foi embebido em resina para realização de cortes ultrafinos da derme reticular, para ser examinado com microscopia eletrônica de transmissão (MET).
ResultadosA MO em pequeno aumento demonstrou homogeneização da derme papilar (fig. 2A). Com outras magnificações, percebe‐se a intensa hialinização da derme papilar, com vasos ectásicos (fig. 2B), ocorrendo também ao redor de glândulas sudoríparas (fig. 2C). Esse depósito dérmico é corado pelo PAS (fig. 2D), e é resistente a diastase.
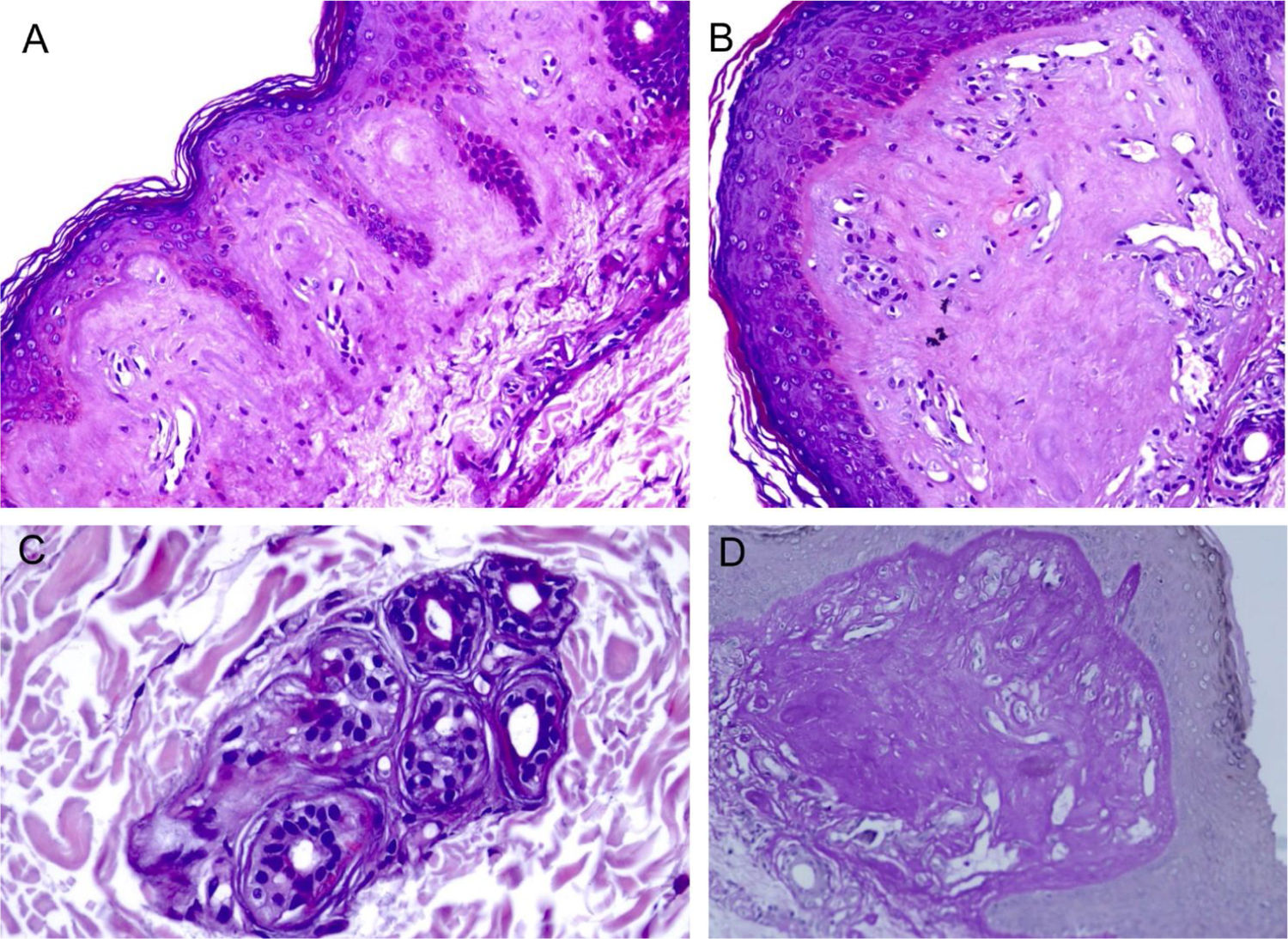
Microscopia óptica. A, Depósito eosinofílico na derme papilar (Hematoxilina & eosina, 150×). (B), Detalhe da derme papilar com homogeneização acentuada e ectasia vascular (Hematoxilina & eosina, 400×). (C), Discreto depósito ao redor de glândula sudorípara (Hematoxilina & eosina, 400×). (D), Coloração PAS demonstrando positividade diastase resistente na derme papilar (ácido periódico de Schiff, 400×).
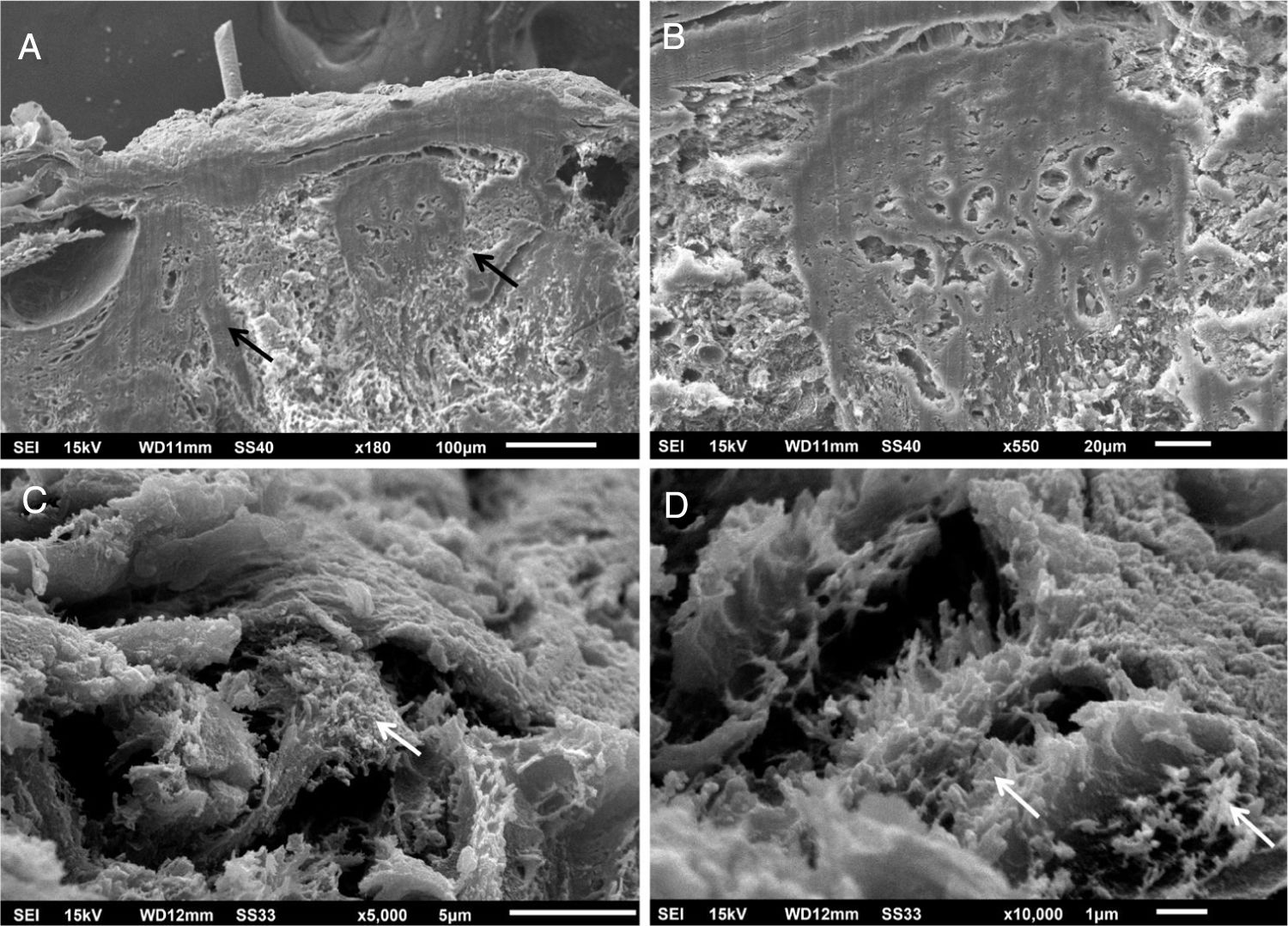
Em pequenos aumentos, de modo semelhante à MO, a MEV demonstrou compactação da derme papilar (fig. 3A), que ficou bem evidente em grandes aumentos (fig. 3B). O exame da derme reticular identificou depósitos fibrilares impedindo a visualização dos feixes de colágeno (figs. 3C e 3D).
A MET demonstrou em pequenos aumentos o depósito de material fibrilar entre os feixes colágenos (fig. 4A) e entre as fibras colágenas (fig. 4B), que apresentavam morfologia normal. Esse depósito foi visto também aderido a fibras elásticas (fig. 4C), e em quantidades maiores em alguns campos examinados deslocando as fibras colágenas (fig. 4D). Estruturas celulares também foram visualizadas e apresentavam o citoplasma preenchido pelo material fibrilar (fig. 5A e 5B). Com grandes aumentos (50.000 ×), a estrutura microfibrilar dos depósitos ficou bem evidente (fig. 5C e 5D).
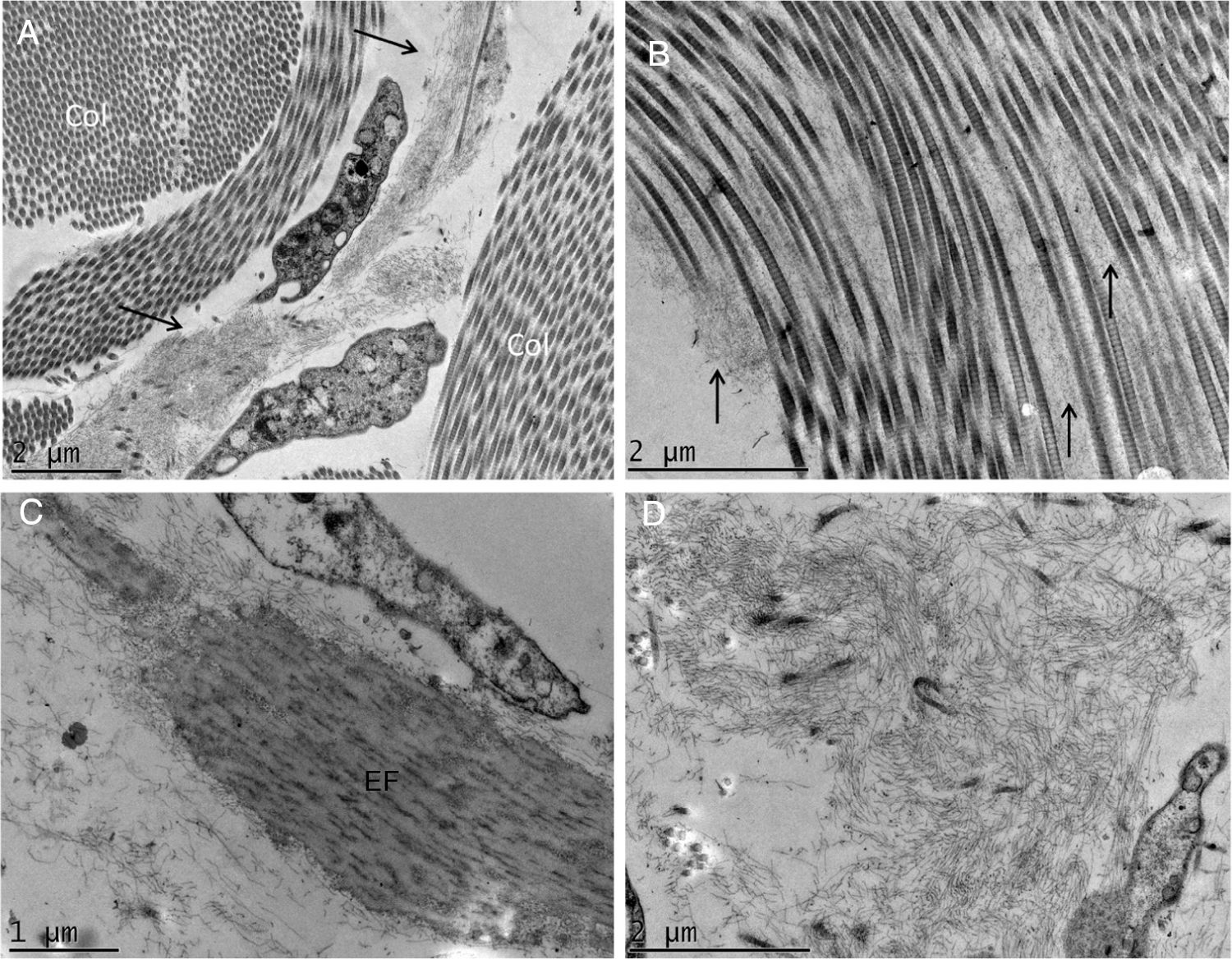
Microscopia eletrônica de transmissão. (A), Depósito de material fibrilar (setas) entre feixes colágenos (col) (15.000×). (B), Depósito fibrilar entre fibras colágenas (setas) (25.000×). (C), Depósito fibrilar ao redor de fibra elástica (FE) (30.000×). (D), Área com deposição importante do material fibrilar com deslocamento das fibras colágenas (25.000×).
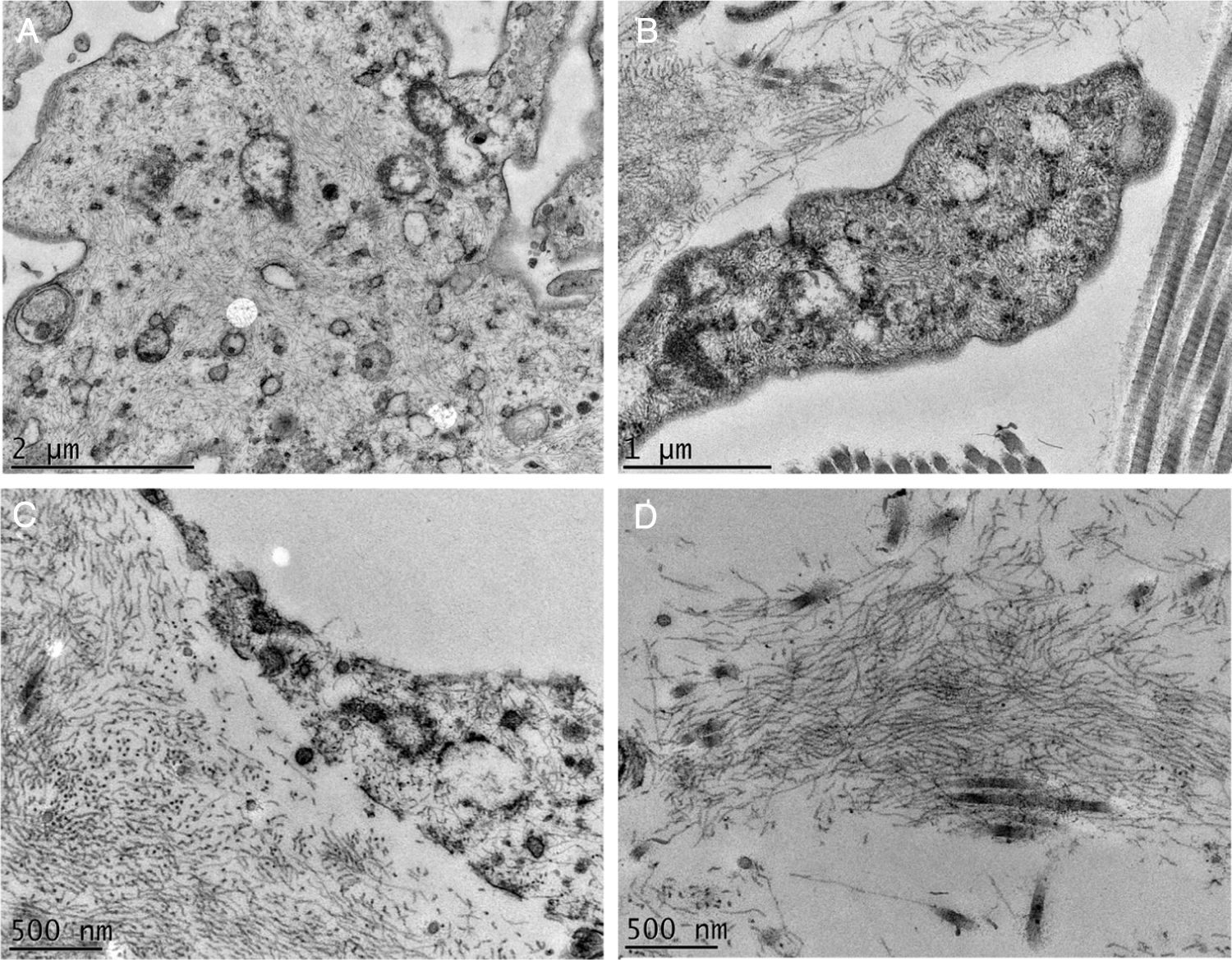
Os achados clínicos e de MO do caso aqui investigado se sobrepõem aos relatados na literatura, sendo bem característica a alteração vista na derme papilar.
Não encontramos relatos com uso da MEV nessa enfermidade, já que essa técnica é raramente utilizada para investigar a derme. Encontramos achados semelhantes à MO na derme papilar, a qual apresenta intensa compactação na imagem tridimensional. Na derme média, os componentes normais, os feixes colágenos, estão cobertos por depósito fibrilar. Com essa técnica, eles aparecem normalmente como filamentos agrupados, formando os feixes, que têm aspecto de cordões.
Na MET, vários autores já relataram o depósito de material filamentar; nossos achados são semelhantes aos de Hashimoto et al., que descreveram filamentos curvados ou anastomosados (formando um aspecto reticular). Esses filamentos também foram vistos dentro de células, sugerindo uma possível fonte de sua origem, como no caso aqui investigado. Alguns autores descreveram os depósitos como granulares.4–6
Estudos bioquímicos sugerem natureza não colagênica dos depósitos, em acordo com nossos achados, já que o colágeno encontra‐se normal – o que fala contra em defeito genético na sua produção, e os depósitos diferirem morfologicamente desse componente dérmico.5
Duplicação de membrana basal epidérmica e vascular também já foram relatadas. Como não evidenciamos essas estruturas em nossos cortes, não temos como fazer comparação com esses achados.7,8
A função exata da proteína mutada ECM1 na lipoidoproteinose não é bem conhecida. Ela é considerada uma glicoproteína da matriz extracelular,3,9 e interage com várias estruturas dérmicas. Há relatos, inclusive, de alteração dérmica vascular na doença, porém é difícil descartar que possam ser eventos secundários.10
Existem quatro variantes da ECM1, o que torna ainda mais difícil a correlação das mutações com os fenótipos – principalmente com as alterações ultraestruturais, já que são poucos os relatos com MET. É possível não só uma certa variabilidade fenotípica, mas também da morfologia ultraestrutural dos depósitos.2 Com a investigação de mais casos, seria possível fazer essas correlações futuramente.
Suporte financeiroNenhum.
Contribuição dos autoresHiram Larangeira de Almeida Jr: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; revisão crítica da literatura; revisão crítica do manuscrito.
Raphael Goveia Rodeghiero: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; revisão crítica da literatura; revisão crítica do manuscrito.
Patrícia Naomi Ando Suzuki: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; revisão crítica da literatura; revisão crítica do manuscrito.
Marília Marufuji Ogawa: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; revisão crítica da literatura; revisão crítica do manuscrito.
Conflito de interessesNenhum.
Como citar este artigo: Almeida Jr HL, Rodeghiero RG, Susuki PN, Ogawa MM. Ultrastructural aspects of the skin in lipoid proteinosis (Urbach‐Wiethe disease). An Bras Dermatol. 2021;96:730–4.
Trabalho realizado na Pós‐Graduação em Saúde e Comportamento, Universidade Católica de Pelotas e Departamento de Dermatologia, Universidade Federal de Pelotas, Pelotas, RS, Brasil.
