







A histiocitose de células de Langerhans é uma doença proliferativa clonal rara, caracterizada pela infiltração de um ou de múltiplos órgãos por histiócitos. Devido à diversidade de sinais e sintomas, o diagnóstico dessa doença é, muitas vezes, tardio. A incidência estimada em adultos é de um a dois casos por milhão, mas provavelmente a doença é subdiagnosticada nessa população. Apresentamos um caso de histiocitose de células de Langerhans disseminada. Destacamos aspectos mais característicos dessa doença rara e heterogênea que, comumente, apresenta‐se como um diagnóstico clínico desafiador.
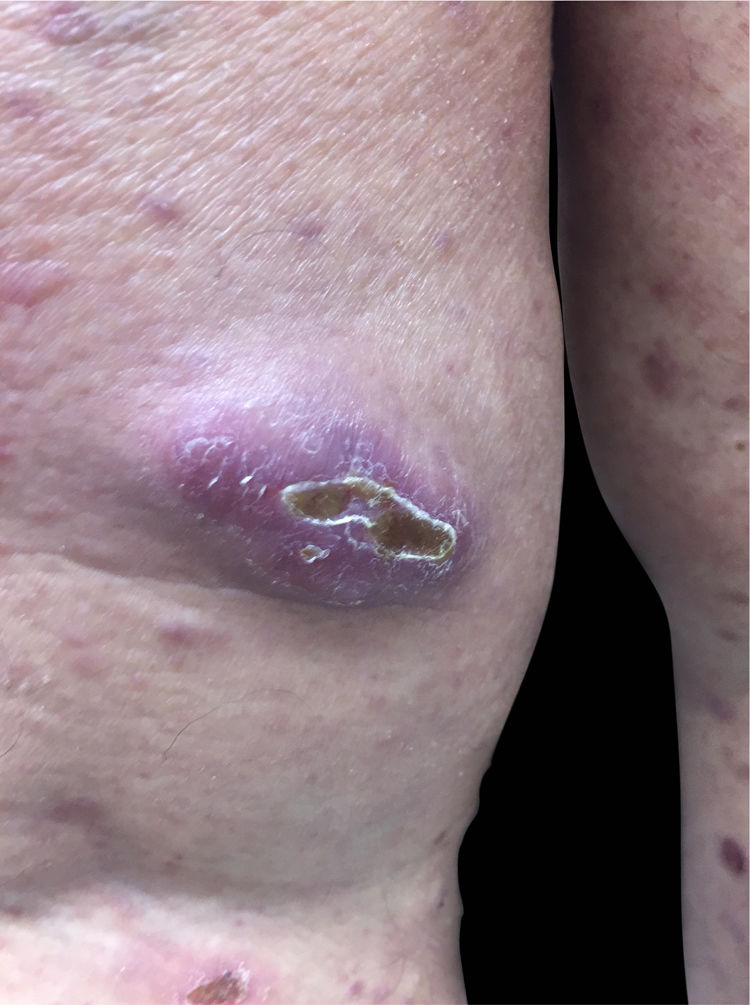
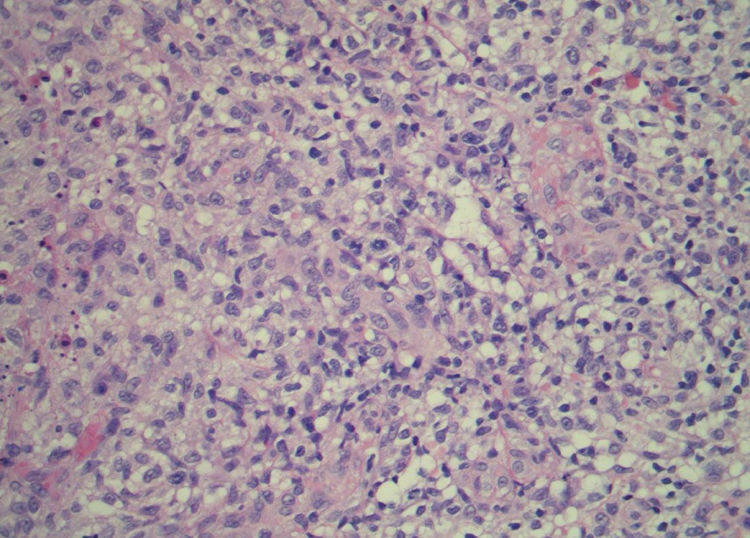
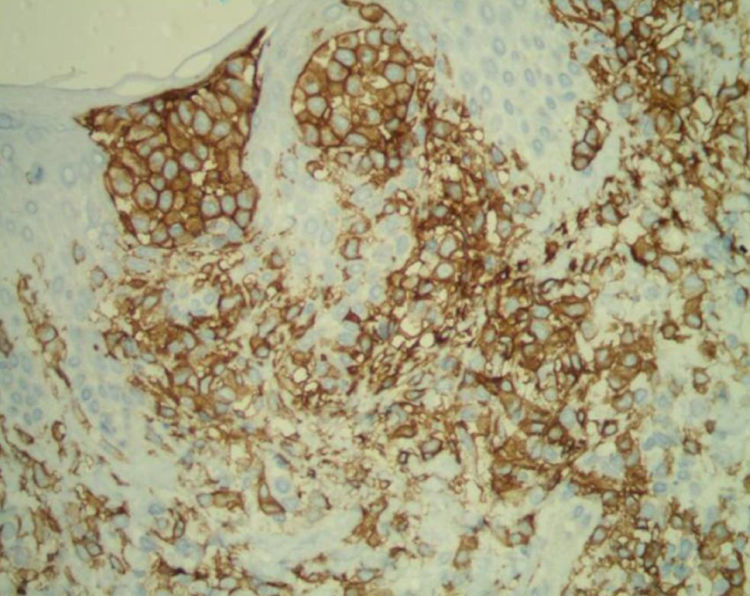
Paciente do sexo feminino, 63 anos de idade, referia manchas avermelhadas pruriginosas e difusas pelo corpo com cerca de seis meses de evolução. Apresentava exames laboratoriais externos com plaquetopenia e anemia e anatomatopatológico sugerindo farmacodermia. Ao exame físico, apresentava múltiplas lesões papulonodulares violáceas por vezes com ulcerações e crostas em extremidades e manchas eritematosas rendilhadas em abdome (figs. 1 e 2). Realizou‐se biópsia cutânea. O exame anatomopatológico evidenciou infiltrado histiocítico em derme papilar e reticular, formando agregados de células de tamanho intermediário, com citoplasma claro e abundante, núcleos por vezes clivados e com pseudofendas (fig. 3). A imuno‐histoquímica demonstrou imunorreatividade para S100, CD1a e langerina, sugerindo, em conjunto com anatomopatológico e história clínica, histiocitose de células de Langerhans (HCL) (fig. 4). A paciente iniciou quimioterapia sistêmica com vimblastina associada à prednisona. Devido à pouca resposta após três ciclos, o tratamento foi substituído por citarabina. A paciente evoluiu com quadro de insuficiência respiratória aguda por provável sepse de foco pulmonar, vindo a óbito.
Discussão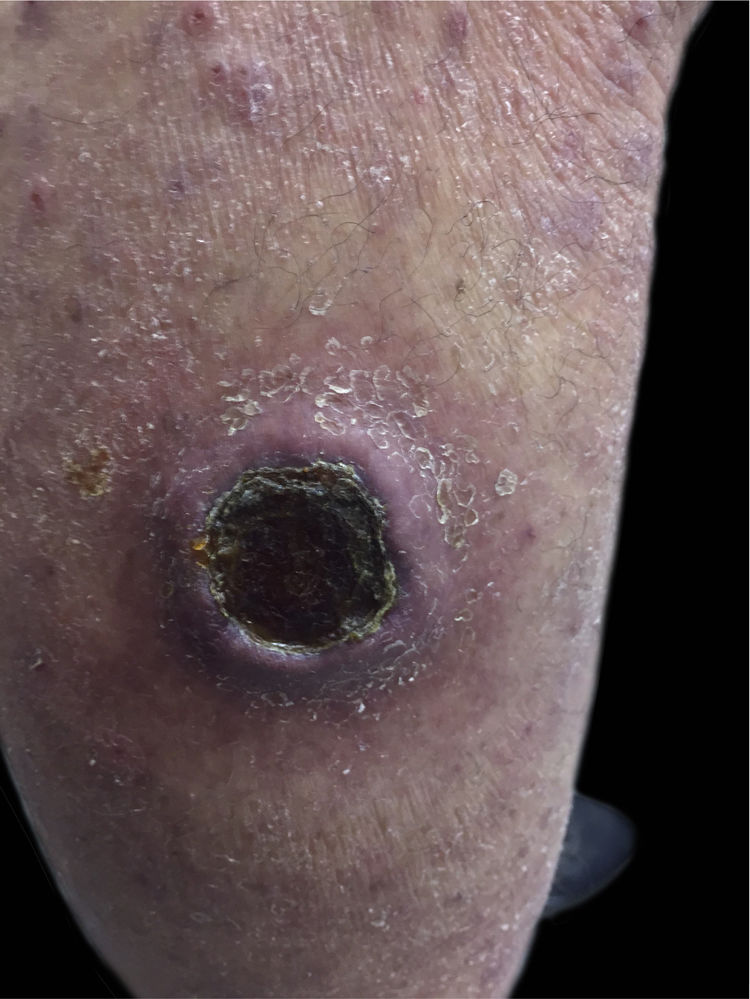
A HCL é uma doença rara e heterogênea. Com a recente descoberta da mutação BRAF‐V600E em uma alta prevalência das HCL (50%?60%), a doença foi reconhecida como neoplásica com marcada inflamação.1,2 Alguns estudos atuais sugerem uma correlação clínica entre a presença da mutação e a recorrência e gravidade da doença.3 Há uma divisão atual em HCL localizada e disseminada. As manifestações clínicas variam muito devido às diferenças entre a idade de início, a taxa de proliferação das células de Langerhans e os tecidos e órgãos envolvidos. O envolvimento ósseo é a forma de apresentação mais comum, tanto no adulto quanto na criança. As erupções cutâneas dessa doença no adulto podem simular outras dermatoses comuns como dermatite seborreia e eczema atópico.4,5 No caso em questão, as lesões eritematosas disseminadas difusas suscitaram inicialmente um diagnóstico clínico de farmacodermia. As lesões cutâneas estão em 40% dos casos associadas à doença multissistêmica; assim, sua presença deve motivar investigação de outros órgãos envolvidos.6 O diagnóstico requer alto grau de suspeição e depende dos achados clínicos e radiológicos associados à histopatologia e à imuno‐histoquímica.4 O padrão‐ouro é a presença da partícula de Birbeck, grânulos presentes no citoplasma das células de Langerhans, na microscopia eletrônica. A principal manifestação imuno‐histoquímica é a presença das proteínas S‐100 e CD1a(+).6,7 O tratamento deve ser individualizado, considerando os órgãos afetados, a extensão da doença e a faixa etária acometida.7 Na doença localizada, como opções terapêuticas estão cirurgia, corticoterapia intralesional, radioterapia local. Quando há doença multissistêmica ou envolvimento de órgãos de risco (baço, fígado, medula óssea e pulmão), está indicada quimioterapia (vimblastina e prednisolona, citarabina, entre outros).2,6 Inibidores de BRAF como vemurafenibe são novas opções terapêuticas.2 Apesar da melhora na sobrevida devido à terapia, a morbidade continua alta para pacientes com HCL, e sequelas permanentes são observadas em cerca de 20%?30% dos pacientes.8 É necessário concentrar o tratamento dessa condição em centros especializados, pois o tratamento deve ser multidisciplinar.3,7
Suporte financeiroNenhum.
Contribuição dos autoresPaulo Henrique Teixeira Martins: Análise estatística; aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Gabriela Dallagnese: Elaboração e redação do manuscrito; revisão crítica da literatura; revisão crítica do manuscrito.
Laura Luzzatto: Participação efetiva na orientação da pesquisa.
Manuela Lima Dantas: Elaboração e redação do manuscrito; revisão crítica da literatura; revisão crítica do manuscrito.
Conflito de interessesNenhum.
Aos preceptores do serviço, à paciente e aos seus familiares.
