













A epidermólise bolhosa adquirida é doença autoimune rara caracterizada pela síntese de autoanticorpos anticolágeno VII, principal componente das fibrilas de ancoragem do hemidesmossoma. A ligação antígeno‐anticorpo suscita uma resposta inflamatória complexa, que culmina na perda da adesão dermoepidérmica na pele e/ou mucosas. Fragilidade cutânea com bolhas, erosões e milia em áreas de trauma caracterizam a forma mecanobolhosa da doença. Já na forma inflamatória da epidermólise bolhosa adquirida predominam placas inflamatórias urticariformes com bolhas tensas, semelhantes ao penfigoide bolhoso, ou lesões mucosas que podem evoluir com cicatrizes permanentes e perda da funcionalidade nas regiões ocular, oral, esofágica, urogenital. Em virtude da similaridade do quadro clínico da epidermólise bolhosa adquirida com outras doenças do grupo penfigoide e com a porfiria cutânea tardia, o diagnóstico da doença é atualmente confirmado principalmente com base na correlação clínica com os achados histopatológicos (clivagem subepidérmica pauci‐inflamatória ou com infiltrado neutrofílico) e na demonstração da presença de IgG anticolágeno VII in situ, por imunofluorescência direta, ou circulante, por meio da imunofluorescência indireta e/ou ELISA. Não há uma terapia específica para a epidermólise bolhosa adquirida, e a resposta ao tratamento é variável, geralmente com remissão completa em crianças e pior prognóstico em adultos com comprometimento mucoso. Corticosteroide sistêmico e imunomoduladores (colchicina e dapsona) são alternativas para o tratamento de formas leves da doença, enquanto formas graves requerem o uso de corticoterapia associada a imunossupressores, imunoglobulina endovenosa e rituximabe (RTX).
Epidermólise bolhosa adquirida (EBA) é dermatose autoimune rara e desencadeada por autoanticorpos contra o colágeno VII, principal componente das fibrilas de ancoragem do epitélio estratificado escamoso. A resultante perda da adesão dermoepidérmica pode se manifestar com leve fragilidade cutânea a estenose mucosa grave.
O primeiro relato de EBA ocorreu em 1895, quando Elliot (1895, apud ROENIGK, 1971, p.1) descreveu dois adultos com fragilidade cutânea adquirida.1 Casos adicionais de EBA foram publicados nos anos subsequentes. No entanto, como o diagnóstico baseava‐se principalmente em características mucocutâneas, não era possível excluir outros diagnósticos diferenciais como porfiria cutânea tardia e penfigoide bolhoso (PB).1 Em 1965, Pass et al. realizaram estudos histoquímicos e sugeriram que a patogenia da EBA estava relacionada com alterações do colágeno.2
Critérios diagnósticos iniciais só foram estabelecidos em 1971 por Roenigk et al.1 A natureza autoimune da EBA foi demonstrada pela presença de depósitos de IgG na zona da membrana basal (ZMB) utilizando estudo de imunofluorescência direta.3 A localização exata dos depósitos de imunocomplexos na lâmina densa foi esclarecida por Yaoita et al.4 e Nieboer et al.5 utilizando imunomicroscopia eletrônica, e uma proteína de 290 kDa – colágeno VII – foi identificada por Woodley et al. em 1984 como o antígeno‐alvo na EBA.6
Relatos mais recentes de pacientes com lesões inflamatórias do tipo PB,7 ou com envolvimento mucoso predominante à semelhança do penfigoide de membranas mucosas (PMM), reforça a necessidade de estudos laboratoriais para demonstrar a presença de autoanticorpos anticolágeno VII8 para confirmação diagnóstica de EBA e diferenciação do grupo penfigoide.
EpidemiologiaA incidência anual da EBA é estimada entre 0,08 a 0,5 casos por milhão de indivíduos,9,10 correspondendo a aproximadamente 5% dos casos de pacientes com anticorpos contra a ZMB.11
A EBA não tem predileção por sexo e geralmente se inicia entre a quarta e quinta décadas de vida.12 Entretanto, indivíduos de qualquer idade podem ser acometidos. Uma metanálise recente revelou que, dentre pacientes com diagnóstico de EBA, 4,6% tinham até 17 anos.13 A EBA na infância ocorre entre 2 semanas de idade a 17 anos.14 A forma clínica inflamatória é a mais frequente e geralmente é acompanhada de lesões mucosas.14 A forma neonatal resultante da passagem placentária de autoanticorpos maternos é descrita na EBA.15
EtiopatogeniaA EBA é doença autoimune que pertence ao grupo das dermatoses bolhosas subepidérmicas. Seu principal alvo antigênico é o colágeno VII (COLVII), localizado na sublâmina densa da ZMB. O COLVII, principal componente das fibrilas de ancoragem, é proteína de 290 kDa que consiste em um domínio central colagênico flanqueado por dois domínios não colagênicos, NC1 e NC2.8 Em pacientes com EBA, a maioria dos autoanticorpos tem como alvo epítopos localizados no domínio NC1, embora reatividade contra os domínios colagênico ou NC2 possa ser detectada em uma minoria dos casos.16 Em geral, tais autoanticorpos são do tipo IgG. Todavia, IgA, IgE e IgM tem sido detectado em alguns pacientes.7
Estudos experimentais sugerem que a susceptibilidade genética à doença está associada em especial ao HLA‐DR2. Mais recentemente, evidências do envolvimento de genes não pertencentes ao complexo MHC foram descritas em modelos experimentais de EBA.7 Estudos mais recentes demonstram o papel protetor da diversidade da microbiota cutânea nas manifestações clínicas da EBA.17,18
Na EBA inflamatória induzida em animais, camundongos deficientes de células T não desenvolvem autoanticorpos específicos contra o COLVII, demonstrando a participação do linfócito T na patogênese da doença. Células T reguladoras também exercem papel protetor para o desenvolvimento da EBA.7 A sensibilização dos linfócitos T CD4+ requer a presença de células apresentadoras de antígeno (APC), cujo fluxo nos linfonodos periféricos é mediado pelo fator estimulador de colônia de granulócitos e macrófagos (GM‐CSF ‐ granulocyte‐macrophage colony‐stimulating) e por neutrófilos.19 Além das APCs, células dendríticas, macrófagos e linfócitos B são necessários para a expansão clonal e diferenciação de células plasmáticas, com subsequente liberação de autoanticorpos contra o COLVII na circulação.7 A síntese de autoanticorpos anti‐COLVII pode ser inibida com o bloqueio da proteína de choque térmico 90 (HSP 90).20
Nos camundongos suscetíveis à doença ocorre polarização para a resposta imune Th1, com aumento da produção de IFN‐γ e IL‐4 nos linfonodos periféricos do sítio de imunização.20
A lesão tecidual inicia‐se pela deposição de autoanticorpos na junção dermoepidérmica por meio da ligação ao epítopo do COLVII, com a ativação do sistema de complemento e liberação de citocinas pró‐inflamatórias7 e quimiotaxia de neutrófilos.21
Neutrófilos ligam‐se ao domínio Fc dos autoanticorpos anti‐COLVII22 e iniciam uma cascata de sinalização que envolve a ativação do receptor órfão relacionado ao retinoide (ROR) α, proteína de choque térmico (HSP) 90, fosfodiesterase 4, fosfatidilinositol‐4,5‐bisfosfato 3‐quinase (PI3K), dentre outras moléculas.7 Desse modo, os neutrófilos são ativados, secretando espécies reativas de oxigênio e proteases.19 Essas substâncias levam à redução das fibrilas de ancoragem, com subsequente formação de bolhas na pele e mucosas.22
Diferentes citocinas têm sido relacionadas com a patogênese da EBA, a exemplo de CXCL1, CXCL2, GM‐CSF e IL‐1α/β, que apresentam expressão aumentada e associam‐se à formação de bolhas na EBA experimental.19 Por outro lado, o papel da IL‐6 ainda não foi totalmente esclarecido. Níveis teciduais e séricos da IL‐6 aumentados correlacionam‐se com a atividade da EBA. No entanto, camundongos que não expressam IL‐6 têm efeito protetor após imunização e não desenvolvem a doença.23
Dados referentes à patogênese da EBA não inflamatória são escassos. Diferentes mecanismos foram propostos, a exemplo de que autoanticorpos ligados ao COLVII comprometem o mesmo, perturbando interações com proteínas da matriz extracelular na ZMB, como colágeno tipo IV e fibronectina.24 Outra possibilidade é de que autoanticorpos interferem diretamente na formação do dímero antiparalelo do COLVII, desestabilizando as fibrilas de ancoragem.25
Aspectos clínicos e classificaçãoA EBA se caracteriza pela presença de bolhas tensas, erosões e fragilidade cutânea. Por terem origem na parte inferior da zona da membrana basal, as bolhas são geralmente bastante tensas e costumam durar vários dias. Podem ter conteúdo límpido ou hemorrágico.7 A presença de milia após a reepitelização das lesões é achado frequente em todas as formas de EBA, e seu encontro é relevante para a inclusão dessa possibilidade diagnóstica, que será confirmada ou excluída pela correlação clínico‐histopatológica‐laboratorial.
A EBA apresenta duas formas clínicas principais: inflamatória e mecanobolhosa (clássica ou não inflamatória). A forma inflamatória é a mais frequente.26,27
EBA mecanobolhosa/ clássica/ não inflamatóriaNa forma mecanobolhosa da EBA a fragilidade cutânea e as lesões vesicobolhosas ocorrem em áreas mais sujeitas a pressão e trauma, principalmente as faces extensoras de regiões acrais (mãos, pés, cotovelos, joelhos, região pré‐tibial). As lesões habitualmente surgem sobre pele normal, sem edema ou eritema. Surgem logo, ou no máximo algumas horas após traumatismos na pele, que podem ser mínimos. Lesões mucosas são frequentes. Outra característica clínica dessa forma é que, durante a evolução, podem surgir milia, cicatrizes atróficas, hiper ou hipopigmentação, distrofia e perda das unhas, alopecia cicatricial, contraturas digitais e estenose do esôfago (fig. 1).27
EBA inflamatória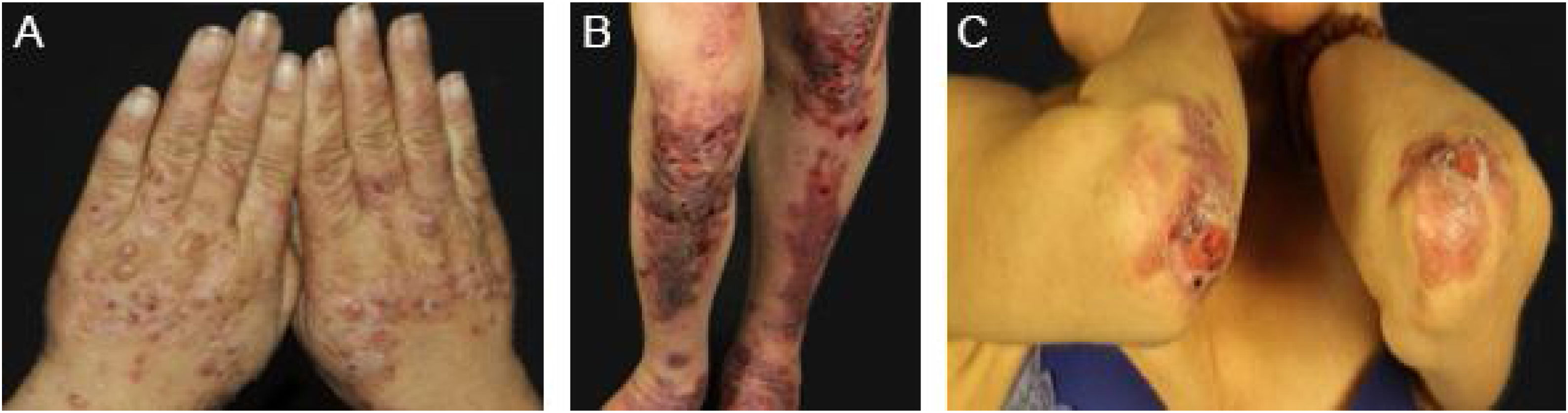
Na forma inflamatória ocorrem lesões em todo o tegumento, não apenas nas áreas mais sujeitas a traumatismos, e a fragilidade cutânea não é tão importante. Pode, portanto, assemelhar‐se a outras dermatoses bolhosas autoimunes (DBA) subepidérmicas, tais como PB, PMM, dermatose bolhosa por IgA linear e penfigoide de Brunsting‐Perry.26–28 O surgimento de cicatrizes e milia durante a evolução é menos frequente do que na EBA mecanobolhosa (fig. 2).27,28
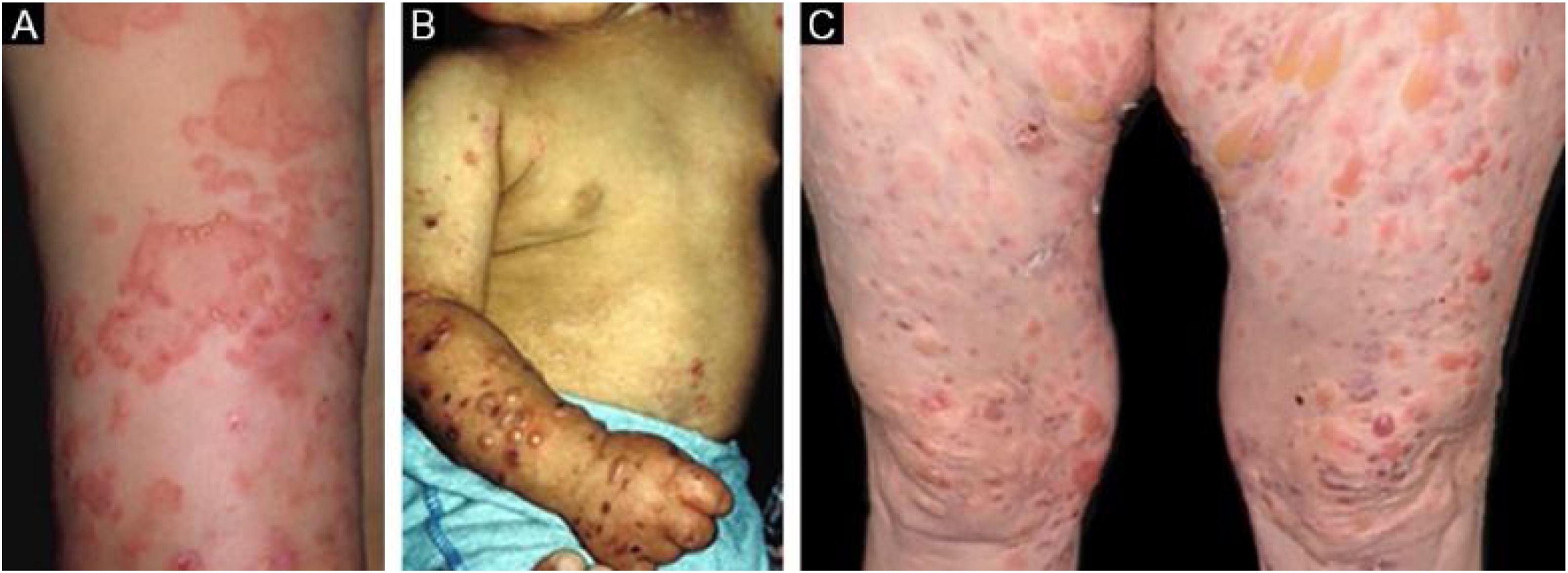
Subtipo mais frequente da forma inflamatória, cursa com lesões vesicobolhosas tensas sobre placas eritêmato‐pruriginosas urticariformes em qualquer parte da pele, inclusive na face, podendo acometer a mucosa oral. Pode haver, à semelhança do PB propriamente dito, áreas onde só se observam placas urticariformes, sem a presença de bolhas. O quadro pode ser indistinguível do PB, tanto clínica quanto laboratorialmente, já que ambos apresentam bolhas subepidérmicas ao exame histopatológico, e na imunofluorescência direta observa‐se o depósito linear de C3 e IgG na zona da membrana basal.28 Algumas vezes os pacientes apresentam, também, lesões sugestivas da EBA mecanobolhosa.29 As lesões podem deixar cicatrizes atróficas e milia após a resolução, ainda que sejam menos frequentes do que na forma clássica.
EBA penfigoide de membranas mucosas‐símileAcomete predominantemente as mucosas, como boca, faringe, esôfago, conjuntiva, ânus, região genital e trato respiratório (traqueia e brônquios).13,27,28,30 Pode ocorrer comprometimento de somente um destes sítios durante período prolongado, dificultando o diagnóstico. Diferentemente do que ocorre no PMM, na EBA PMM‐símile as bolhas mucosas podem ser duradouras e estar intactas no momento do exame físico. As lesões cicatriciais (cicatrizes atróficas, sinéquias e estenoses) são idênticas às que ocorrem no PMM. Essas lesões cicatriciais têm consequências às vezes mais leves na mucosa oral, genital e anal, mas podem causar prejuízo funcional importante no esôfago, laringe, traqueia, brônquios e conjuntiva, podendo levar a simbléfaro, triquíase e até perda da visão.31,32 A estenose de esôfago geralmente ocorre em sua porção superior e gera disfagia, perda de peso, desnutrição e até infecção pulmonar, por aspiração de alimentos.30,32–35 Lesões importantes no trato respiratório podem levar a perfuração de septo nasal, estenose de faringe, laringe e, mais raramente, traqueia e brônquios, podendo levar a asfixia.36 A abordagem multidisciplinar é mandatória nesses casos (fig. 3).
EBA dermatose por IgA linear‐símile (EBA IgA)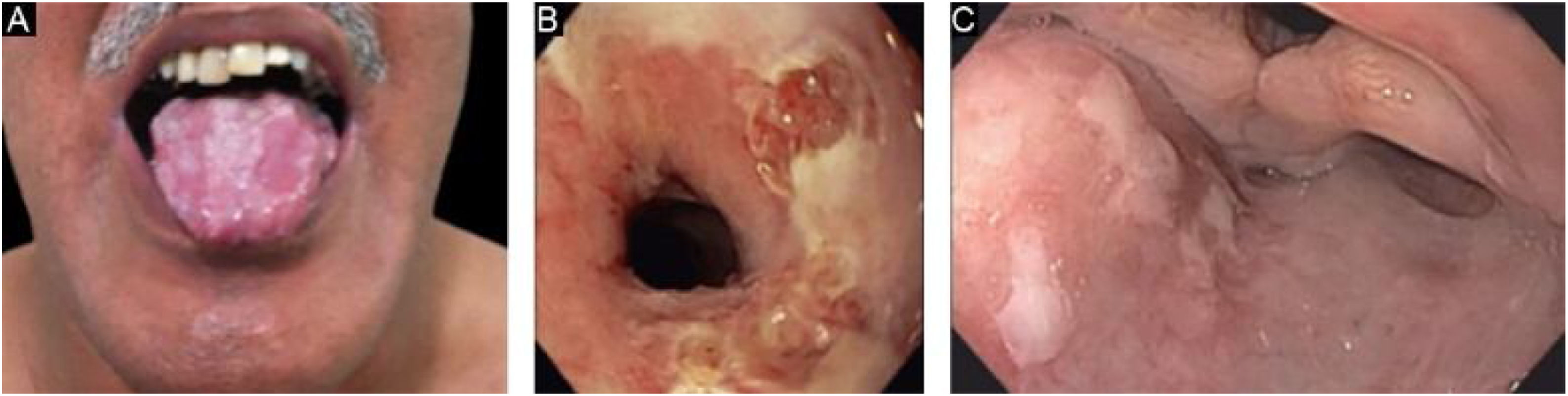
Caracteriza‐se pelo depósito linear de IgA na ZMB, no seu lado dérmico. Assemelha‐se clinicamente à dermatose bolhosa por IgA linear, com a presença de lesões anulares (em roseta), poucas cicatrizes e milia. Raramente cursa, no entanto, com formação de cicatrizes mucosas, inclusive quadro ocular importante.27,37 Pode apresentar resposta terapêutica à dapsona, à semelhança do observado na dermatose bolhosa por IgA linear.26,37
EBA Brusting‐Perry PMM‐símileAs lesões são apenas na pele e restritas à região da cabeça, pescoço e ombros. Pode cursar com erosões persistentes e cicatrizes atróficas.27,28,38,39.
Há poucas séries de casos de EBA publicadas, que analisam a frequência relativa de cada uma das formas clínicas da EBA.26,31 Importante destacar que os critérios diagnósticos utilizados podem variar entre as publicações. As duas formas consideradas mais frequentes são a EBA mecanobolhosa e a PB‐símile. A forma clínica pode variar, no mesmo paciente, ao longo do tempo (p. ex., da forma PB‐ símile para a mecanobolhosa).7,12 A EBA também pode ocorrer em crianças, e nessa população a forma inflamatória também parece ser a mais frequente. A mucosa oral é mais acometida, e a resposta ao tratamento parece ser melhor do que na população adulta.
A EBA tem, de modo geral, impacto bastante importante na qualidade de vida dos pacientes. Isso pode ser mensurado pelos escores específicos para dermatoses bolhosas autoimunes, o ABQOL (Autoimmune Bullous disease Quality of Life) e TABQOL (Treatment‐Based Autoimmune Bullous disease Quality of Life).40,41
Doenças associadas à EBADiversas doenças sistêmicas têm sido encontradas em associação com a EBA, tais como amiloidose, tireoidite, síndrome da endocrinopatia múltipla, artrite reumatoide, fibrose pulmonar, leucemia linfoide crônica, timoma e diabetes mellitus. No entanto, a maior parte desses relatos são isolados.7,12 A única associação indiscutível da EBA é com a doença inflamatória intestinal (DII), particularmente doença de Crohn. Essa associação é observada em cerca de 25% dos pacientes com EBA nos Estados Unidos; é mais rara em outros países e provavelmente decorre da presença de colágeno tipo VII na ZMB da parede do intestino grosso. Na maior parte dos pacientes, DII precede o surgimento da EBA.26,42,43
DiagnósticoComo a EBA apresenta características mucocutâneas polimórficas com gravidade variável, pode confundir‐se com qualquer outra dermatose bolhosa autoimune subepidérmica, como PB, PMM, doença bolhosa por IgA linear e lúpus eritematoso sistêmico bolhoso (LESB). O diagnóstico baseia‐se não somente na correlação clínico‐patológica, mas também requer a demonstração da presença de autoanticorpos IgG in situ e/ou circulantes contra o colágeno VII.16
HistopatologiaOs achados histopatológicos na EBA variam conforme o tipo e duração da lesão.16 Na EBA clássica ou mecanobolhosa, pode‐se observar clivagem subepidérmica com edema papilar e infiltrado inflamatório escasso (fig. 4).7,44 Por outro lado, a EBA não clássica ou inflamatória apresenta intenso infiltrado inflamatório com neutrófilos, eosinófilos e linfócitos na derme papilar/superficial (fig. 4).44 Lesões tardias geralmente apresentam cistos de queratina (milia) e fibrose dérmica.7
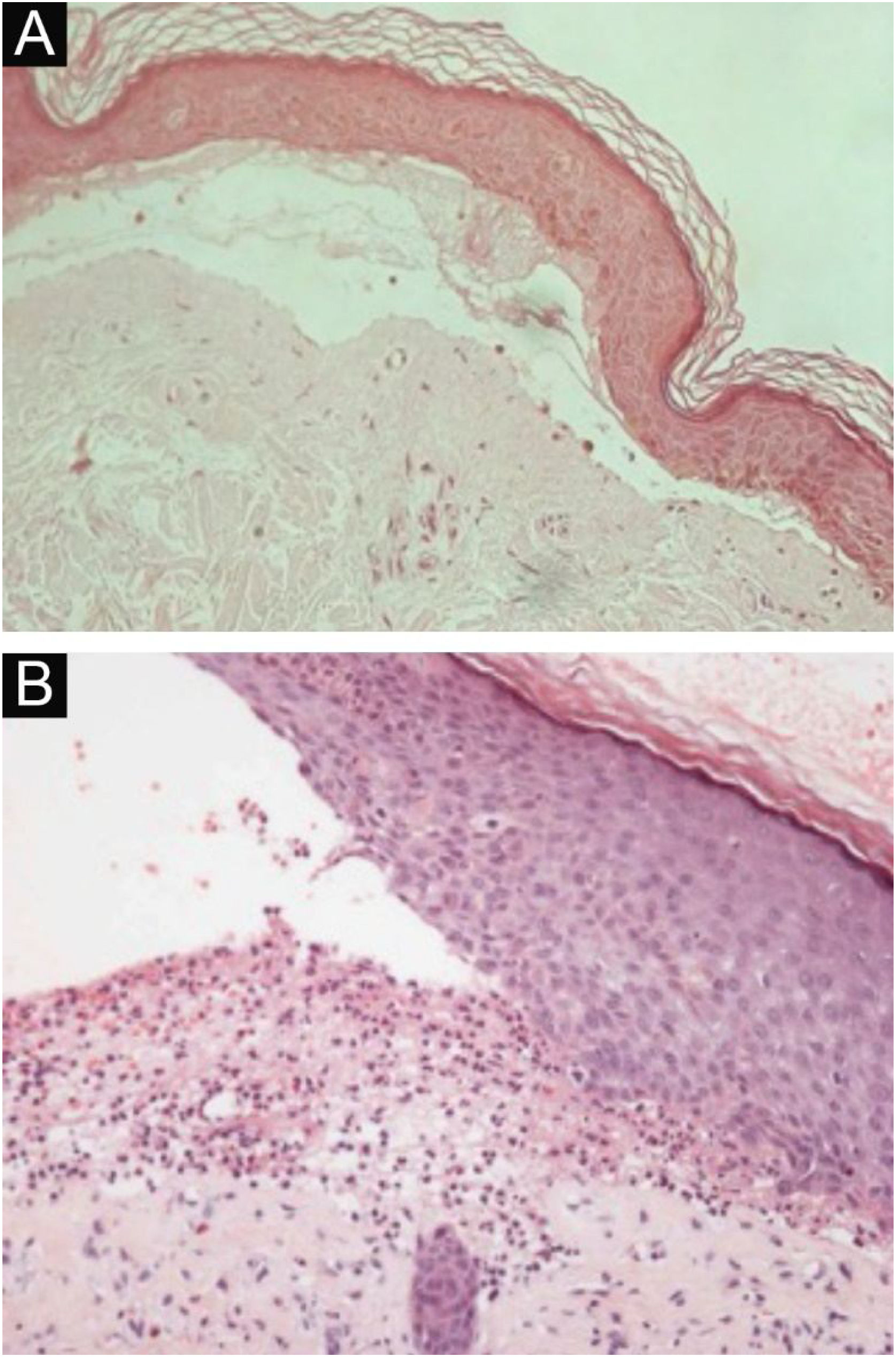
Exame anatomopatológico da epidermólise bolhosa adquirida com coloração por hematoxilina‐eosina. (A), Forma mecanobolhosa, com clivagem subepidérmica com infiltrado inflamatório escasso (200×). (B), Forma inflamatória, com clivagem dermoepidérmica e rico infiltrado inflamatório perivascular neutrofílico (400×).
Depósitos lineares de IgG e C3 na ZMB estão presentes na pele perilesional8 em 93% dos pacientes.16 Esses achados não são exclusivos de EBA e também podem ser encontrados em outras DBA subepidérmicas.45 Fluorescência com anti‐C3 é observada em 89% dos casos, seguida por anti‐IgG em 79% dos casos;45 IgA (47%) e IgM (21%) são menos frequentemente observados (fig. 5).44,45 Depósitos exclusivos de IgA podem ser encontrados em 2,4% dos casos correspondentes à EBA IgA.13
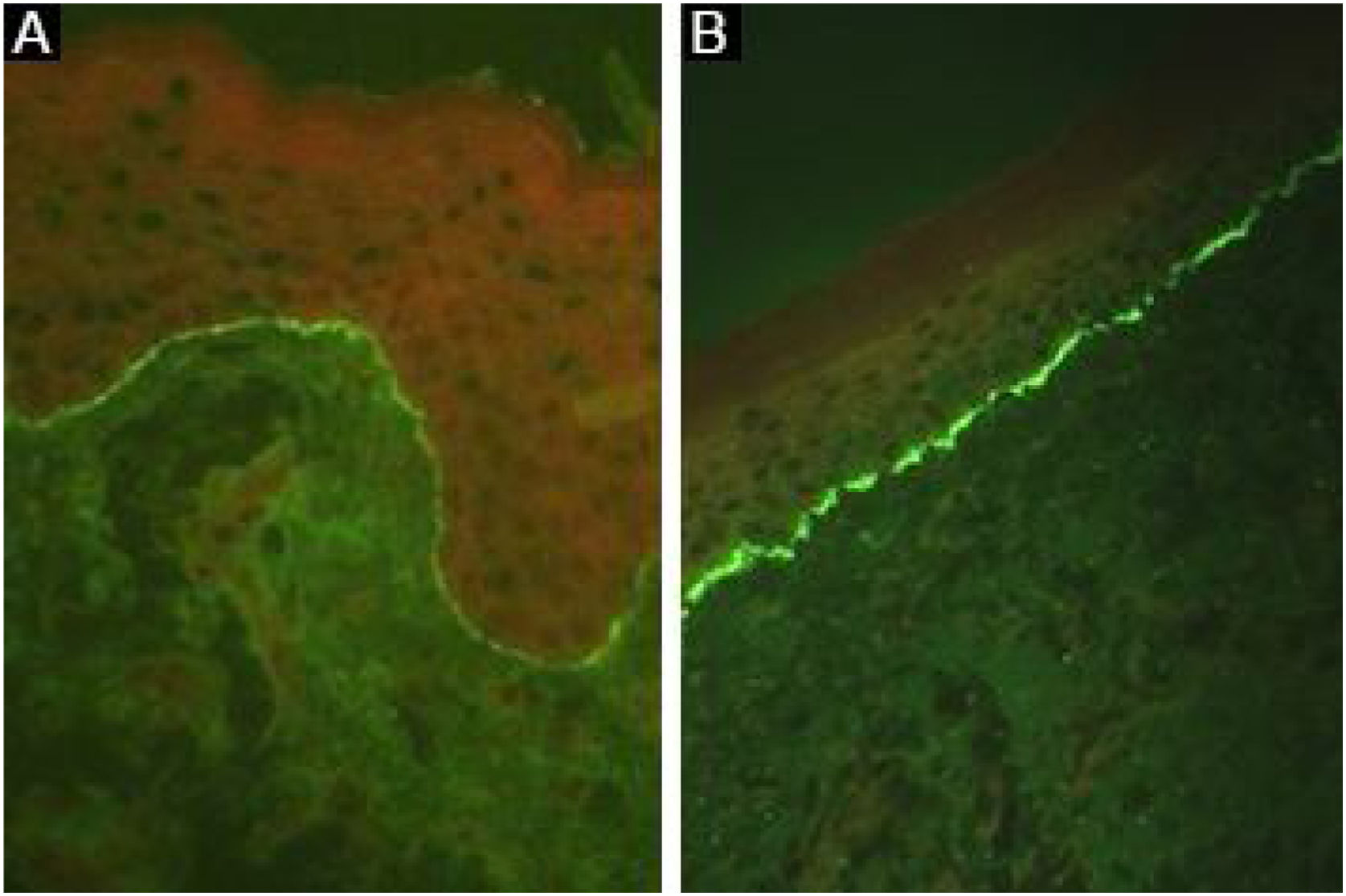
Já foi descrito que a análise do padrão de depósitos de imunocomplexos pode aumentar a sensibilidade da IFD:16 uma fluorescência de padrão u‐serrilhado sugere a presença de autoanticorpos ligados ao colágeno VII das fibrilas de ancoragem, enquanto a fluorescência de padrão n‐serrilhado indica o reconhecimento dos antígenos situados acima da lâmina densa, como BP180, p200, laminina 332 e laminina γ1.8 No entanto, a análise de padrões requer treinamento para avaliação adequada e não se encontra amplamente disponível.
A técnica de salt‐split skin pode ser realizada no fragmento obtido de uma área lesada da pele do paciente, revelando fluorescência do lado dérmico da clivagem. No entanto, tal análise é mais comumente realizada por imunofluorescência indireta, uma vez que a incubação do fragmento de pele lesada com NaCl 1M pode danificar o espécime e prejudicar a visualização dos depósitos de imunocomplexos, reduzindo a sensibilidade do exame.46
Imunomicroscopia eletrônicaDemonstração de depósitos de IgG, C3 e IgA47 nas fibrilas de ancoragem abaixo da lâmina densa8,16 utilizando imunomicroscopia eletrônica direta permanece como a técnica padrão‐ouro para o diagnóstico de EBA.44 Como esse método encontra‐se indisponível na maioria das instituições, estudos adicionais são recomendados para confirmar o diagnóstico de EBA (fig. 6).

Critérios diagnósticos de epidermólise bolhosa adquirida (EBA).8 IFD, imunofluorescência direta; IFI, imunofluorescência indireta; NC, domínio não colagênico; IEM, imunoeletromicroscopia eletrônica; FOAM, fluorescence overlay antigen mapping – mapeamento de antígenos por sobreposição de imunofluorescência.
O fluxograma para o diagnóstico de EBA baseia‐se na correlação entre o exame dermatológico e os resultados dos estudos complementares, uma vez que não há achado clínico ou laboratorial que isoladamente possibilite a confirmação diagnóstica. Atualmente se sugere considerar EBA como hipótese diagnóstica nos casos em que há fragilidade cutânea com formação de cicatriz e milia (forma mecanobolhosa) ou placas urticariformes com bolhas (forma inflamatória). O quadro clínico correlaciona‐se com os achados do exame anatomopatológico: a forma mecanobolhosa apresenta uma clivagem subepidérmica pauci‐inflamatória, enquanto na forma inflamatória observa‐se uma dermatose bolhosa dermoepidérmica rica em neutrófilos. O caráter autoimune da doença pode ser confirmado pelo exame de imunofluorescência direta, que possibilita a visualização de depósitos de imunocomplexos principalmente por IgG, seguida por C3, IgA e IgM. Além disso, a pesquisa de autoanticorpos circulantes pode ser realizada por meio de diferentes métodos diagnósticos como imunoeletromicroscopia eletrônica indireta, ELISA, imunofluorescência indireta (substrato de epitélio escamoso estratificado normal ou biochip) e Western blot. Quando os exames previamente mencionados resultam negativos ou inconclusivos, a pesquisa de autoanticorpos in situ pode ser realizada por imunoeletromicroscopia eletrônica direta, padrão do serrilhado da imunofluorescência direta ou mapeamento de antígenos por sobreposição de imunofluorescência (FOAM). No entanto, como essas técnicas encontram‐se mais disponíveis para pesquisa, na prática clínica costuma‐se utilizar a imunofluorescência direta ou indireta com salt‐split skin como método para demonstrar os depósitos de imunocomplexos no lado dérmico da clivagem, onde se localiza o colágeno VII.
Essa técnica compara a localização de um antígeno conhecido da ZMB com os imunocomplexos depositados na pele perilesional obtida de pacientes com EBA,7 utilizando imunomarcações com diferentes cores fluorescentes.8
Imuno‐histoquímicaFragmentos embebidos em formol e fixados em parafina obtidos da pele lesada de pacientes com EBA são corados com anticolágeno IV, que se localiza na lâmina densa. Como o nível de clivagem na EBA ocorre abaixo da lâmina densa, o colágeno IV será positivo no teto da bolha.8
Autoanticorpos circulantesAutoanticorpos circulantes na EBA podem ser demonstrados em cerca de 50% dos pacientes e correlacionam‐se com a gravidade da doença.48 A sensibilidade varia de acordo com a metodologia utilizada, conforme descrito a seguir.
Imunofluorescência indiretaA presença de IgG anticolágeno VII circulante pode ser demonstrada utilizando amostras de soro obtidas de pacientes com EBA. Após incubação com amostra de pele normal, esôfago de macaco ou bexiga de rato,16,44 os depósitos de imunocomplexos na ZMB são observados como fluorescência linear em até 37% dos pacientes.44 Já a positividade com anti‐IgA costuma ocorrer em títulos baixos (1:2 até 1:320)37 e é observada em 2,3% dos casos.13
A sensibilidade pode ser aumentada até 74,7% utilizando a técnica de salt‐split,8 na qual a pele normal humana é clivada no nível da lâmina lúcida com NaCl 1M, aumentando a exposição de antígenos do hemidesmossoma. Como o colágeno VII permanece no lado dérmico da clivagem, a incubação com soro de pacientes com EBA produzirá uma fluorescência linear no assoalho da bolha (fig. 7).48
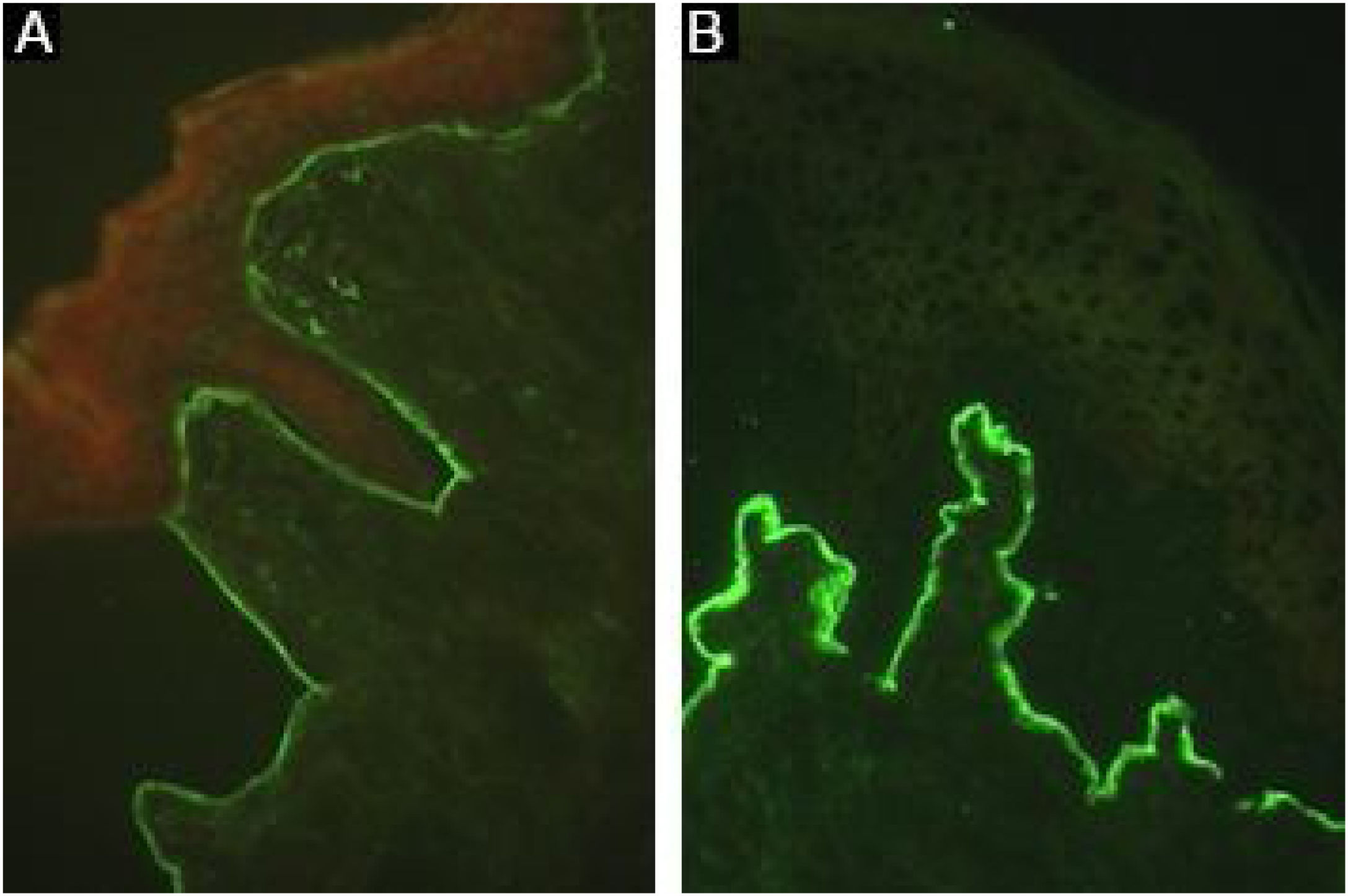
Fragmentos de pele exibindo menor expressão de colágeno VII, obtidos de pacientes com epidermólise bolhosa distrófica, podem também ser utilizados como substrato. Em pacientes com EBA, a incubação de soros pré‐selecionados positivos na pele humana normal resultará negativa ou com fluorescência menos intensa após incubação com amostra de pele de epidermólise bolhosa distrófica.8
A análise da subclasse de IgG pode fornecer informações adicionais para a diferenciação entre EBA do LESB, uma vez que o colágeno VII é o antígeno‐alvo em ambas as doenças; IgG1 e IgG4 são observadas na EBA, enquanto IgG2 e IgG3 são geralmente detectados no LESB.49
ELISASistemas de ELISA comercialmente disponíveis contendo os epítopos não colagênicos (NC) 1 e/ou NC2 podem ser utilizados para demonstrar e quantificar a presença de autoanticorpos IgG contra o colágeno VII.16 A sensibilidade foi descrita entre 20‐98%44,50 e varia conforme o tipo de proteína recombinante NC utilizada e a seleção de soro sabidamente positivo de pacientes com EBA.8 A positividade correlaciona‐se com a atividade da doença,50 porém não com a forma de apresentação clínica (envolvimento mucoso vs. cutâneo).7
Western blotDiferentes epítopos de colágeno VII podem ser obtidos a partir de proteínas recombinantes e extratos de células ou tecidos para produzir uma banda de 290 kDa e/ou 145 kDa.44,47 A sensibilidade varia entre 20‐80%, de acordo com a fonte de colágeno VII e o tipo de domínio NC utilizado.44 Resultados falso‐negativos podem ocorrer em virtude de alterações conformacionais nos epítopos durante o preparo das proteínas recombinantes.51
BiochipIgG anticolágeno VII circulante pode ser detectada após incubação do soro com células humanas transfectadas com o domínio NC1. A formação de imunocomplexos é revelada utilizando um anticorpo anti‐humano secundário marcado com fluoresceína, com sensibilidade e especificidade semelhantes aos descritos para estudos com ELISA.8
Exames complementaresO dermatologista exerce papel fundamental na coordenação do seguimento multidisciplinar dos pacientes com EBA. O rastreamento de acometimento mucoso na EBA é crucial para avaliar a presença de lesões ativas e/ou sequelas e determinar a melhor abordagem terapêutica de acordo com a gravidade da doença (tabela 1).16,47 Também se recomenda avaliar a associação com doença de Crohn, assim como de comorbidades.47
Rastreamento recomendado para pacientes com epidermólise bolhosa adquirida: avaliação do comprometimento mucoso16,47
| Envolvimento mucoso | Nasal e orofaríngeo | Nasofibroscopia |
| Esofágico | Endoscopia digestiva alta | |
| Ginecológico | Vulvoscopia | |
| Colposcopia | ||
| Ocular | Exame oftalmológico |
A EBA mecanobolhosa pode ser clinicamente indistinguível da porfiria cutânea tarda (PCT), uma vez que ambas apresentam fragilidade cutânea com a formação de bolhas e cicatrizes em áreas suscetíveis a trauma. Hipertricose, disfunção hepática, aumento da ferritina e dos níveis de porfirinas podem ser observados na PCT e contribuem para seu diagnóstico.52 O exame anatomopatológico caracteriza‐se pela presença de festonamento das papilas dérmicas e espessamento da parede vascular, especialmente com a coloração de PAS (periodic acid‐schiff). Depósitos homogêneos de IgG, IgM, IgA na zona da membrana basal e na parede dos vasos são os achados mais comuns à IFD na PCT e auxiliam na diferenciação da EBA.53
Quando os pacientes apresentam fragilidade cutânea com início precoce (congênita) ou têm antecedente familiar de epidermólise bolhosa (EB), faz‐se necessário excluir o diagnóstico de epidermólise bolhosa distrófica hereditária.47 Na EB congênita, a IFD resultará negativa enquanto o imunomapeamento revelará uma diminuição ou ausência de colágeno VII.54
A EBA inflamatória pode se manifestar com placas urticariformes pruriginosas e bolhas, como no PB.7 O exame anatomopatológico no PB pode auxiliar na diferenciação diagnóstica, pela presença de espongiose eosinofílica ou de uma dermatite bolhosa subepidérmica com eosinófilos. A IFD tem como achados mais frequentes a presença de depósitos lineares de C3 e de IgG na ZMB e a IFI com a técnica de salt‐split skin possibilita diferenciar da EBA, uma vez que no PB ocorre fluorescência no lado epidérmico ou epidérmico e dérmico da clivagem.55
Pacientes com EBA podem apresentar envolvimento predominantemente mucoso, com resolução da inflamação formando cicatrizes nos olhos, boca, nariz, faringe, laringe, esôfago, uretra e/ou ânus. A sequela subsequente assemelha‐se à observada no penfigoide de membranas mucosas ou na doença bolhosa por IgA linear.47 A histopatologia do PMM é comumente caracterizada por uma clivagem dermoepidérmica pauci‐inflamatória, enquanto na doença bolhosa por IgA linear verifica‐se uma dermatite bolhosa subepidérmica neutrofílica. Na IFD, o PMM pode ser indistinguível das outras formas de penfigoide e EBA. Por esse motivo, a IFI com a técnica de salt‐split skin é um exame que contribui para a diferenciação diagnóstica: enquanto se observa fluorescência do lado epidérmico da clivagem na maioria dos casos de PMM, na EBA a fluorescência é observada do lado dérmico da clivagem. Entretanto, em casos de PMM anti‐p200 e antilaminina 332, também se observa fluorescência no lado dérmico da clivagem.56 Os casos de dermatose bolhosa por IgA linear e EBA com depósito exclusivo de IgA linear na ZMB por IFD podem ser diferenciados pela IFI com a técnica de salt‐split skin. Na dermatose bolhosa por IgA linear, a fluorescência ocorre no lado epidérmico da clivagem, enquanto na EBA essa fluorescência é verificada no lado dérmico da clivagem.55
O LESB é outro potencial diagnóstico diferencial quando bolhas tensas surgem principalmente em áreas fotoexpostas de pacientes do sexo feminino com fotossensibilidade, e que geralmente evoluem sem a formação de cicatrizes ou milia.57 Os achados dos exames histopatológico e de IFD e IFI são semelhantes no LESB e na EBA, uma vez que em ambas as doenças há formação de anticorpos anticolágeno VII. Os achados laboratoriais que confirmam o diagnóstico de lúpus sistêmico, como a positividade de autoanticorpos antinucleares e comprometimento extracutâneo, a exemplo do renal, hematológico, articular e neurológico, são critérios que auxiliam na diferenciação entre LESB e EBA.57
TratamentoO tratamento da EBA tem por objetivo controlar a atividade da doença e prevenir recorrências e sequelas permanentes. O controle da doença é definido como cessação do surgimento de novas lesões e cicatrização das preexistentes.58 É crucial avaliar o grau de comprometimento mucocutâneo, a presença de sequelas, a idade do paciente e as comorbidades para determinar a escolha terapêutica.
A prevenção de novas lesões envolve a educação do paciente para: (1) evitar danos utilizando roupas com tecido suave e curativos não aderentes, (2) realização de limpeza adequada das lesões com água e sabonete, (3) interromper a ingestão de alimentos que possam provocar traumas adicionais à mucosa oral durante a mastigação e deglutição, como bebidas quentes e produtos ácidos, ásperos/crocantes, (4) aderir ao tratamento proposto e às visitas de acompanhamento para realização de ajustes precoces da terapia se necessário.
Reavaliação frequente do comprometimento mucoso por equipe multidisciplinar especializada é também importante para o diagnóstico precoce de novas lesões, prevenção da formação de cicatrizes e introdução do tratamento adequado.16 Sequelas podem necessitar de tratamento cirúrgico incluindo plástica ocular, remoção de sinéquia nasal, uretral e ginecológica, dilatação esofágica ou gastrostomia se houver formação de estenose grave e traqueostomia em caso de comprometimento laríngeo com redução do lúmen e obstrução da via aérea.59
Em virtude da raridade dessa dermatose bolhosa autoimune e manifestação clínica diversa, faltam estudos controlados randomizados a respeito do tratamento da EBA. As recomendações atuais baseiam‐se em relatos e séries de casos, bem como na opinião de especialistas reunidos em consensos e diretrizes nacionais e internacionais.47,59–61 A maioria sugere que o tratamento de escolha deve ser programado de acordo com a gravidade da doença, que pode ser objetivamente avaliada pelo Índice de Área de Doença do Penfigoide Bolhoso (Bullous Pemphigoid Disease Area Index ‐ BPDAI) ou subjetivamente pelo grau de comprometimento mucoso e risco de sequelas e limitações funcionais de longo prazo.47,59 Assim, manifestações leves exclusivamente cutâneas poderiam ser tratadas com corticosteroides sistêmicos e imunomoduladores, enquanto que o comprometimento ocular, laríngeo, esofageano e uretral requer o uso de corticoterapia associada a imunossupressores e/ou RTX e IVIg para o controle da doença (fig. 8).60
Corticosteroide sistêmico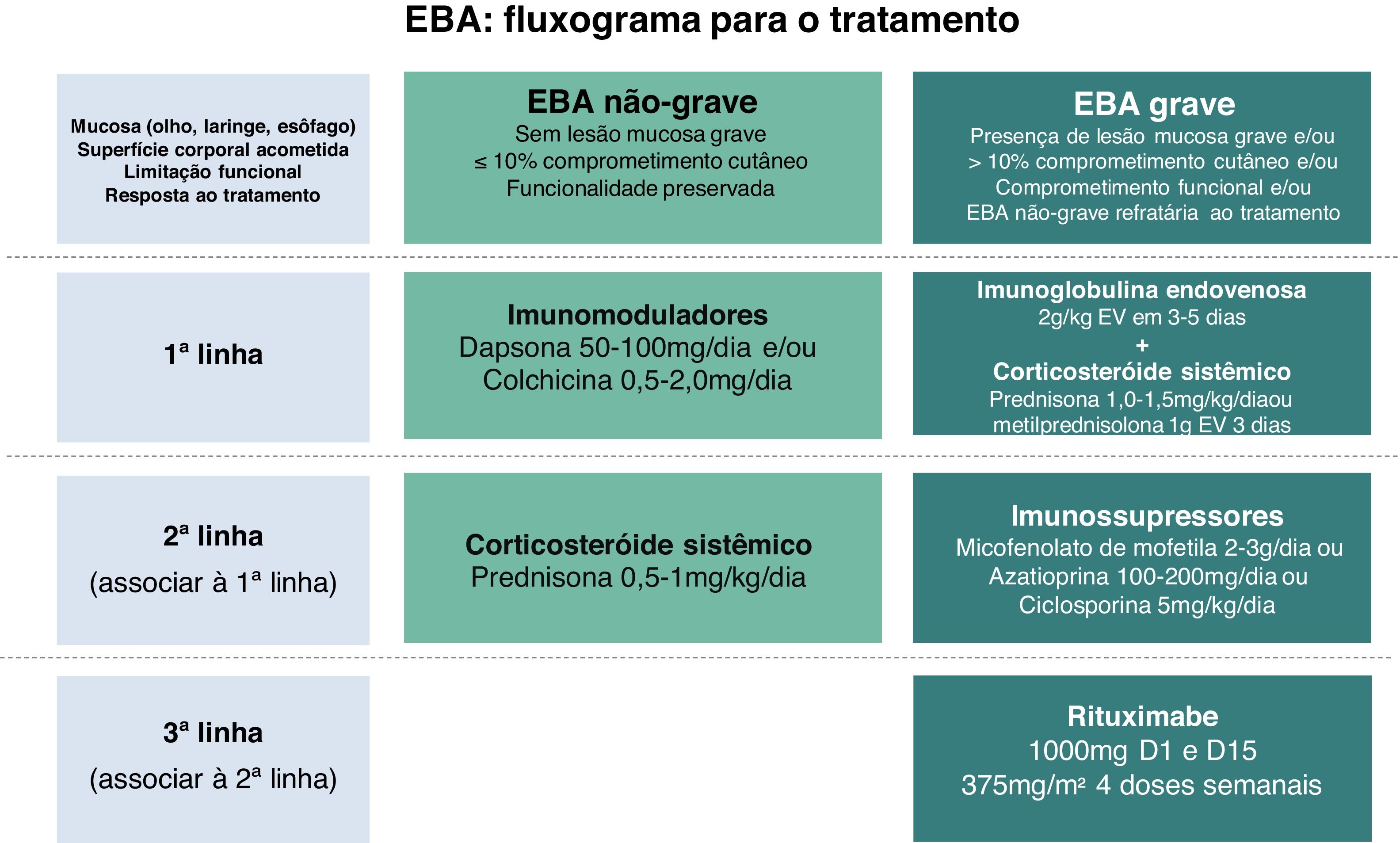
Corticosteroide oral, endovenoso ou em pulsoterapia é considerado a primeira escolha de tratamento da EBA, embora as evidências atuais sugiram que promova melhor controle principalmente da forma inflamatória da doença.62 O mecanismo de ação preciso dos corticosteroides na EBA não foi totalmente esclarecido. Sabe‐se que a corticoterapia induz neutrofilia e reduz a liberação de citocinas e os níveis de linfócitos, eosinófilos e monócitos.63 Modelos experimentais demonstram que a metilprednisolona atua nos neutrófilos, inibindo a fosforilação de quinases como p38 MAPK, ERK 1/2 e Akt. A inativação dessas vias resulta na redução da síntese de radicais livres de oxigênio e da degranulação de neutrófilos, com subsequente diminuição da clivagem subepidérmica.64
A dose inicial recomendada varia de 0,5 mg/kg/dia em casos leves a 1,5 mg/kg/dia em casos graves. Para pacientes apresentando disfagia, soluções de corticosteroide oral podem ser mais efetivas e toleráveis do que comprimidos. Como a corticoterapia sistêmica prolongada relaciona‐se a diversas complicações incluindo ocular (catarata, glaucoma), metabólica (obesidade, diabete, hipertensão dislipidemia, osteoporose, síndrome de Cushing), osteoarticular (necrose asséptica de cabeça de fêmur), imunossupressores, imunomoduladores e RTX são utilizados por seu efeito poupador de corticosteroide.63
DapsonaA dapsona (DDS) 25‐150 mg/dia reduz a quimiotaxia de neutrófilos e apresenta efeito poupador de corticosteroide.8 A DDS pode mesmo ser utilizada em monoterapia com controle adequado da atividade da EBA mediada por IgA7 e em pacientes pediátricos.8,62 A resposta à doença geralmente é observada dentro de duas semanas de tratamento.62 Em virtude dos potenciais efeitos adversos como anemia hemolítica, meta‐hemoglobinemia, agranulocitose e DRESS (drug reaction with eosinophilia and systemic symptoms; reação à droga com eosinofilia e sintomas sistêmicos), faz‐se necessária monitoração laboratorial frequente. Também se recomenda a avaliação dos níveis de glicose‐6‐fosfato desidrogenase, pois níveis baixos correlacionam‐se com hemólise.
ColchicinaEBA leve pode ser controlada utilizando colchicina 1‐2 mg/dia em monoterapia ou combinada com corticoterapia sistêmica com poucos efeitos adversos como diarreia dose‐dependente.7 A colchicina é vantajosa em pacientes em que o uso de imunossupressores deve ser evitado, uma vez que promove a inibição da quimiotaxia de neutrófilos e o aumento da prostaglandina E2.65
CiclosporinaA ciclosporina (CyA) 4‐9 mg/kg/dia foi raramente utilizada como tratamento adjuvante na EBA, apesar de ter sido relatada melhora em 11/11 pacientes.16 Nefrotoxicidade, assim como outros potenciais efeitos adversos como hipertensão, hipertricose, dislipidemia e cefaleia7 podem limitar a utilização da CyA.
Micofenolato de mofetilaO micofenolato de mofetila (MMF) 2‐3 g/dia tem sido utilizado com sucesso em combinação com corticoterapia sistêmica para promover remissão da doença e mesmo em monoterapia após desmame completo do corticosteroide.66 A maioria dos dados a respeito da eficácia e segurança do MMF originam‐se de estudos incluindo pacientes com pênfigo vulgar ou PB,7 e seu mecanismo de ação específico na EBA permanece desconhecido. Como MMF atua na síntese de purina dos linfócitos,67 a resultante depleção de células B possivelmente reduz a síntese de autoanticorpos contra o colágeno VII.66 Náusea, diarreia, hepatite e linfopenia foram relatadas como efeitos adversos comuns.67 Estudos adicionais são necessários para melhor esclarecer o papel do MMF no tratamento da EBA.
AzatioprinaA azatioprina (AZA) 2‐3 mg/kg/dia pode ser combinada com corticoterapia sistêmica na EBA moderada a grave.7 O efeito imunossupressor envolve a inibição da síntese de ácido nucléico e proteínas, com depleção de células mononucleares e linfócitos.67 A atividade da tiopurina metiltransferase interfere no metabolismo da AZA, e a determinação de seus níveis pode ser útil para avaliação da dose do tratamento e prevenção de efeitos adversos dose‐dependentes como hepatite e leucopenia.7,67 Efeitos adversos idiossincráticos adicionais incluem náusea, pancreatite, diarreia.7 Avaliação pré‐tratamento e exames laboratoriais regulares são recomendados (tabela 2).
Principais fármacos utilizados no tratamento da epidermólise bolhosa adquirida7–9,25–33,63,65,67,71–76
| Fármaco (dose) | Principais mecanismos de ação | Principais efeitos adversos | Avaliação laboratorial |
|---|---|---|---|
| Corticosteroide sistêmico (prednisona 0,5‐1,0 mg/kg/dia)a ou (prednisona 1,0‐1,5 mg/kg/dia)b ou (metilprednisolona 1 g/dia IV por 3 dias)b | Inibição de citocinas, citopenias (eosinófilos, linfócitos, monócitos), neutrofilia | Ocular (catarata, glaucoma), metabólico (obesidade, diabetes, hipertensão, dislipidemia, osteoporose, síndrome de Cushing), osteoarticular (necrose asséptica da cabeça do fêmur) | Hemograma completo, enzimas hepáticas, função renal, glicemia de jejum, hemoglobina glicada colesterol total e frações, triglicérides, densitometria |
| Dapsonaa (50‐100 mg/dia) | Ação antineutrofílica, com redução da quimiotaxia de neutrófilos | Anemia hemolítica, meta‐hemoglobinemia, agranulocitose, DRESS (drug reaction with eosinophilia and systemic symptoms) | Glicose‐6‐fosfato desidrogenase, hemograma completo, enzimas hepáticas, lactato desidrogenase, reticulócitos, bilirrubina total e frações |
| Colchicinaa (0,5‐2,0 mg/dia) | Ação antineutrofílica, com inibição da quimiotaxia de neutrófilos e o aumento da prostaglandina E2 | Neutropenia, diarreia, desconforto abdominal | Hemograma, enzimas hepáticas, função renal |
| Ciclosporinab (5 mg/kg/dia) | Inibição da calcineurina fosfatase, causando depleção de células T e macrófagos, e da ativação de células natural killer, células T e células apresentadoras de antígenos | Nefrotoxicidade, hipertensão, hipertricose, dislipidemia, cefaleia | Hemograma completo, enzimas hepáticas, função renal, colesterol total e frações, triglicérides |
| Micofenolato de mofetilab (2‐3 g/dia) | Inibição da síntese de purina, causando depleção de linfócitos | Náusea, diarreia, hepatite, linfopenia | Hemograma completo, enzimas hepáticas, função renal, sorologias para hepatite B, C, HIV |
| Imunoglobulina endovenosa (2 g/kg EV em 3‐5 dias) | Depleção de autoanticorpos por redução da meia‐vida de imunoglobulinas | Cefaleia, dor torácica, febre, dispneia, mialgia, náusea, vômito, diarreia, taquicardia, eritema, anafilaxia, insuficiência renal aguda, tromboembolismo, meningite asséptica, neutropenia, anemia hemolítica | Hemograma completo, função renal, enzimas hepáticas, dosagem de imunoglobulinas séricas (atentar para deficiência de IgA, pelo risco aumentado de anafilaxia) |
| Rituximabeb (1.000mg D1 e D15 ou 375 mg/kg/m2, quatro doses semanais) | Anticorpo monoclonal quimérico anti‐CD20 que induz depleção de linfócitos B por indução de apoptose, ativação do complemento e citotoxicidade | Febre, náusea, vômito, angioedema, broncoespasmo, anafilaxia, infecção, reativação de hepatite B, angina, arritmia, insuficiência cardíaca, síndrome coronariana | Hemograma completo, enzimas hepáticas, função renal, sorologias para hepatite B, C, HIV |
A literatura sobre o uso de ciclofosfamida (CyP) oral (50‐100 mg/dia) ou em pulsoterapia endovenosa (500‐1000 mg/m2) é rara.7,61 A combinação de CyP e pulsoterapia com corticosteroide tem sido relatada como tentativa de controle da doença em pacientes com comprometimento mucoso grave. CyP inibe a síntese de DNA e induz apoptose. Porém, mielossupressão, cistite hemorrágica, infertilidade e carcinogênese61 limitaram a utilização da CyP, principalmente após o advento do RTX.67
MetotrexatoO metotrexato (MTX) 20‐25 mg/semana é utilizado em combinação com corticoterapia sistêmica como um agente poupador de corticosteroide, uma vez que inibe a síntese de ácido nucléico e a ativação linfocitária.68 Poucos relatos a respeito do uso do MTX na EBA foram publicados, portanto não é possível avaliar adequadamente a eficácia do fármaco para controle da doença.7 Efeitos adversos comuns incluem alopecia, citopenias, desconforto abdominal e hepatotoxicidade.68 Recomenda‐se avaliação laboratorial antes da introdução de MTX e durante o tratamento (tabela 2).
Imunoglobulina endovenosaImunoglobulina endovenosa (IVIg) 2 g/kg em 3‐5 dias consecutivos é alternativa de tratamento para EBA grave e recalcitrante,7 uma vez que reduz os autoanticorpos patogênicos.69 Segundo metanálise recente sobre o tratamento de EBA, somente o tratamento com IVIg esteve associado à remissão completa na EBA não mecanobolhosa.13 No entanto, pacientes com EBA tratados com IVIg necessitam de 16‐22 ciclos para atingir controle da doença, o que limita seu uso em virtude do alto custo do tratamento.58 IVIg também é útil em combinação com RTX para reduzir o risco infeccioso após a infusão de anti‐CD20.69 O efeito adverso mais comum da IVIg é a cefaleia.7
Plasmaférese/imunoadsorçãoA remoção de autoanticorpos circulantes em combinação com o tratamento com RTX tem sido relatada para o controle de casos de EBA recalcitrante.7 Porém, plasmaférese e imunoadsorção não se encontram amplamente disponíveis e, por isso, têm sido utilizados em poucos casos.
RituximabeA remissão completa da EBA tem sido relatada com o uso do RTX, anticorpo monoclonal quimérico anti‐CD20. Os esquemas de tratamento mais utilizados são de 1.000 mg/infusão no D1 e D15 (protocolo artrite reumatoide) ou 375 mg/m2/semana por quatro semanas (protocolo linfoma).7
Uma revisão da literatura realizada em 2018 avaliou o tratamento da EBA com RTX em monoterapia ou combinado com IVIg ou imunoadsorção. Dentre 16 pacientes tratados com RTX em monoterapia com 500 ou 1.000mg no D1 e D15, 13 responderam inicialmente (três com resposta parcial, três com resposta completa, cinco com remissão da doença, um óbito por pneumonia no D21, e uma perda de seguimento) e três não tiveram resposta. A combinação de RTX 375 mg/m2/semana por quatro semanas com IVIg 2 g/kg induziu melhora clínica e controle da doença em 5/5 pacientes, porém devido à persistência de lesões cutâneas em 4/5 pacientes, tratamento de manutenção com IVIg mensal foi necessário. RTX (protocolo linfoma) e imunoadsorção foram usados em cinco pacientes, dos quais dois atingiram controle da doença. Como a filtração do plasma com remoção de imunoglobulinas e reinfusão não é uma técnica amplamente disponível, não é possível avaliar a eficácia da imunoadsorção na EBA até o momento.70
Pacientes com EBA comumente apresentam episódios de exacerbação durante a evolução da doença, por vezes com refratariedade ao tratamento convencional. Uma metanálise identificou 1.159 casos de EBA relatados de 1971 a 2016 e avaliou qual fármaco induziu resposta completa uma vez utilizada em monoterapia.13 A maioria dos pacientes portadores de EBA fez uso de múltiplos medicamentos, em virtude da ineficácia de tratamentos anteriores. Apesar das limitações do estudo incluindo sua natureza retrospectiva, o escasso número de relatos de casos e a heterogeneidade de tratamentos e desfechos, os autores concluíram que imunoglobulina intravenosa e RTX foram associados à remissão clínica da doença. A análise de subgrupo, entretanto, demonstrou que a resposta ao tratamento entre as variantes da EBA foi distinta: a imunoglobulina intravenosa foi associada à remissão completa (RC) na EBA mecanobolhosa, enquanto nenhum tratamento foi associado à RC na EBA não mecanobolhosa.
Perspectivas futuras no tratamento da EBADescobertas a respeito da fisiopatogenia da doença possibilitaram a caracterização de potenciais alvos terapêuticos incluindo: imunobiológicos que inibem a ativação do sistema complemento (anticorpos antifração do receptor gama – FcR); proteínas que diminuem a expressão de interleucinas pró‐inflamatórias como IL‐6, e estimulam a síntese de citocinas anti‐inflamatórias como IL‐4 e IL‐10; moléculas e vias que reduzem a quimiotaxia e ativação de neutrófilos (LTB4, p38 MAPK, GM‐CSF).7,19 Os novos fármacos para o tratamento da EBA têm sido estudados e desenvolvidos com base em modelos experimentais animais e, portanto, sua eficácia e segurança em humanos ainda precisa ser elucidada.
Evolução e prognósticoA EBA é doença crônica de alta morbidade devido ao seu potencial cicatricial (figs. 9 e 10). Formação de estenoses e sinéquias secundárias à atividade mucosa da doença podem ocorrer de maneira subclínica.30 Seu reconhecimento tardio aumenta o potencial de complicações graves com impacto significativo na qualidade de vida.77 Em geral, o prognóstico e a resposta ao tratamento da EBA são melhores em crianças.78
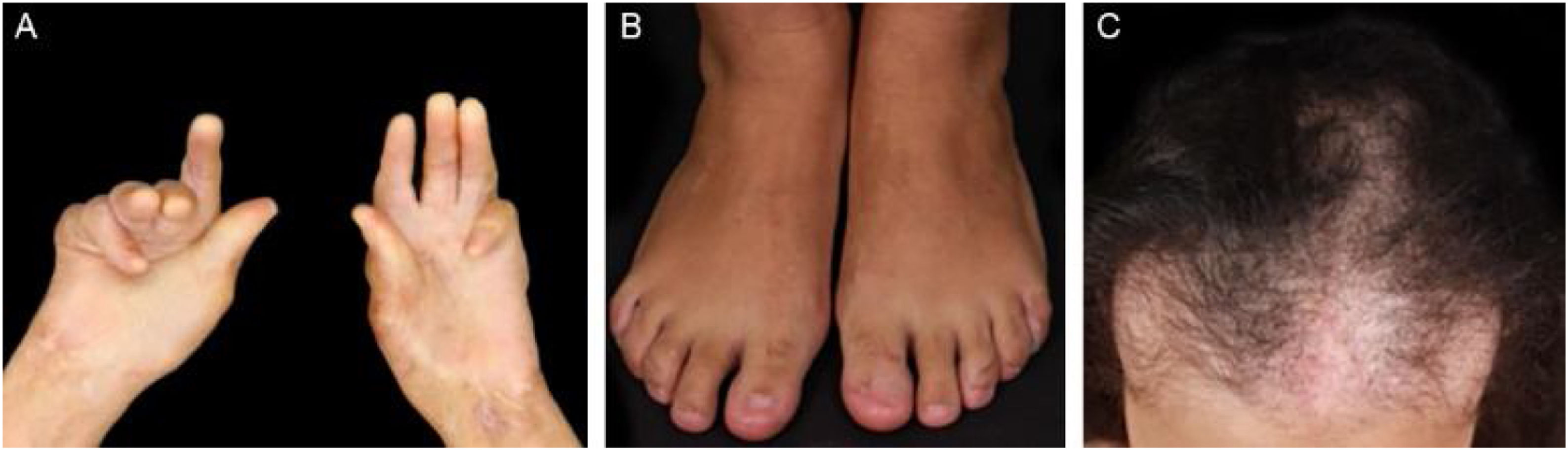
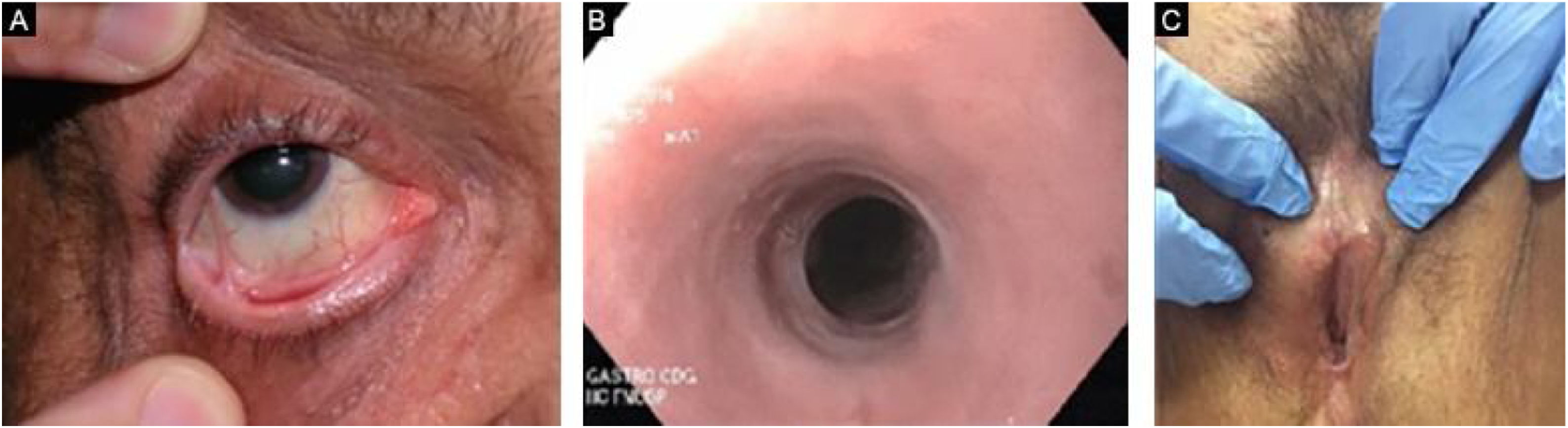
Estudo multidisciplinar com quatro portadores de EBA demonstrou que 50% dos pacientes já apresentam sinéquias nas vias aéreas antes do surgimento de sintomas significativos.30 Outro estudo incluindo pacientes com EBA77 mostrou que 5/12 pacientes apresentaram complicações mucosas, dentre elas, estenose esofágica (2/12), sinéquias laríngeas (2/9), simbléfaro e triquíase (2/12) e deformidade em mãos (3/12).
Embora dados referentes a fatores prognósticos da EBA sejam escassos na literatura, acredita‐se que a evolução e prognóstico da doença estejam associados a gravidade no momento do diagnóstico e a resposta ao tratamento proposto.7 Correlação entre gravidade e atividade da doença e níveis séricos de autoanticorpos anti‐COLVII também foi descrita.50,79
Em estudo retrospectivo com 30 pacientes,26 após um ano de seguimento, observou‐se resposta parcial em 20,8% dos pacientes e 33,3% evoluíram com resposta completa. O tempo para atingir a remissão e o percentual de resposta entre as formas mecanobolhosa e inflamatória de EBA foi semelhante. Os autores utilizaram o conceito de remissão para EBA com base na adaptação das definições descritas pelo Comitê Internacional de Pênfigo.80
Suporte financeiroNenhum.
Contribuição dos autoresDenise Miyamoto: Revisão crítica da literatura; elaboração e redação do manuscrito; aprovação da versão final do manuscrito; revisão crítica do manuscrito.
Juliana Olivieri Gordilho: Revisão crítica da literatura; elaboração e redação do manuscrito; aprovação da versão final do manuscrito; revisão crítica do manuscrito.
Claudia Giuli Santi: Revisão crítica da literatura; elaboração e redação do manuscrito; aprovação da versão final do manuscrito; revisão crítica do manuscrito.
Adriana Maria Porro: Revisão crítica da literatura; elaboração e redação do manuscrito; aprovação da versão final do manuscrito; revisão crítica do manuscrito.
Conflito de interessesNenhum.
Como citar este artigo: Miyamoto D, Gordilho JO, Santi CG, Porro AM. Epidermolysis bullosa acquisita. An Bras Dermatol. 2022;97:409–23.
Trabalho realizado no Departamento de Dermatologia, Hospital das Clínicas, Faculdade de Medicina, Universidade de São Paulo e da Escola Paulista de Medicina, Universidade Federal de São Paulo, São Paulo, SP, Brasil.
