






A histiocitose de células de Langerhans (HCL) é neoplasia inflamatória de células precursoras mieloides na qual ocorre o acúmulo de células dendríticas especializadas em diferentes órgãos.1 Relatamos um caso raro de HCL com apresentação multissistêmica.
Paciente masculino, 2 meses de idade, apresentando desde o nascimento lesões eritêmato‐purpúricas esparsas pelo corpo. Após curto período de aparente melhora, as lesões recidivaram. O paciente não apresentava manifestações sistêmicas.
Ao exame físico, observavam‐se pápulas angiomatosas encimadas por crostas hemáticas e algumas lesões, hipocrômicas, de aspecto brilhante, acometendo inclusive região palmoplantar e cavidade oral (fig. 1).
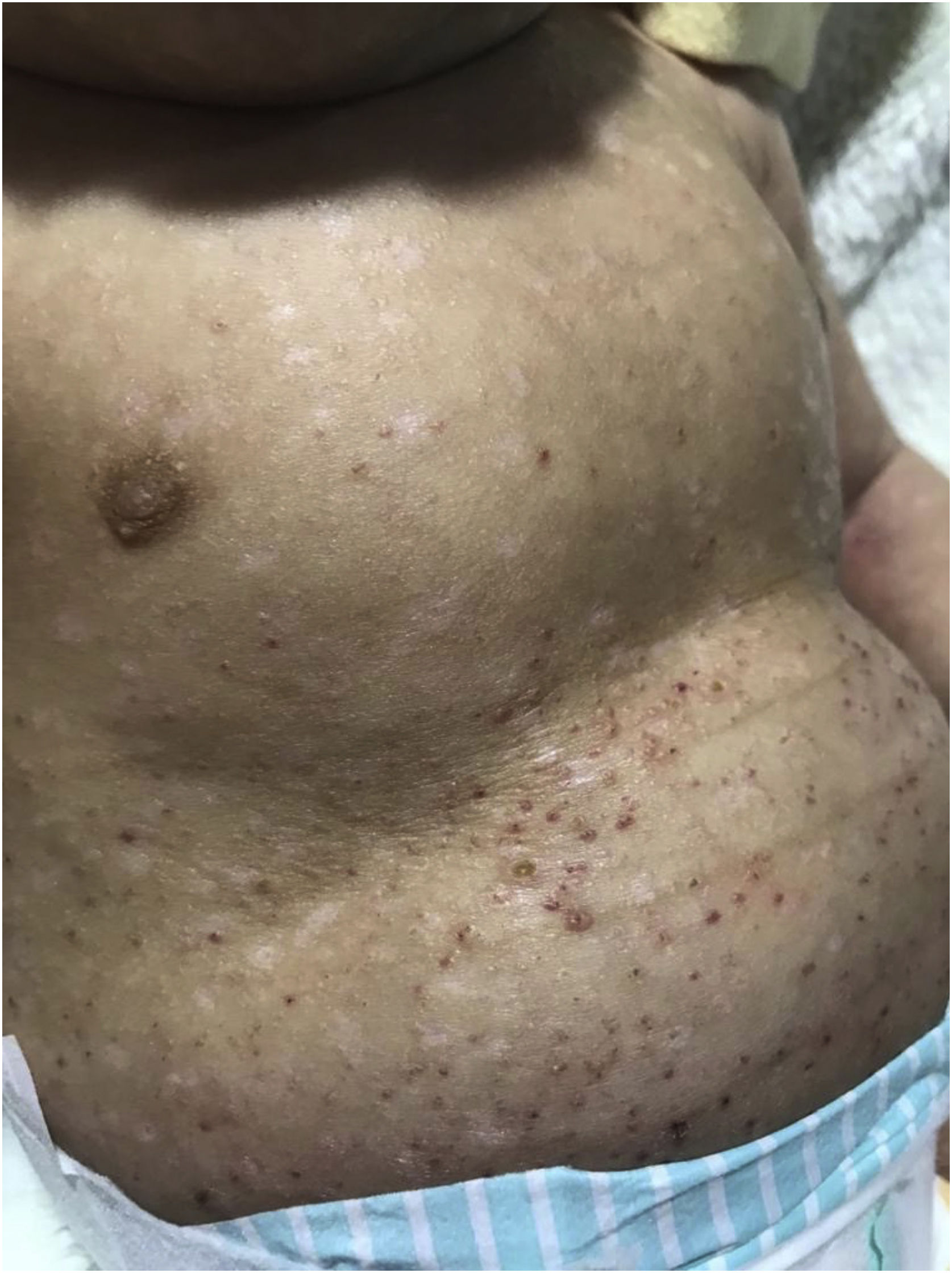
Diante disso, foram aventadas as hipóteses diagnósticas de HCL, citomegalovirose congênita (CMV), leucemia cútis e imunodeficiência combinada grave. Realizada biópsia, solicitadas sorologias (HIV, CMV, rubéola, toxoplasmose, VDRL) que resultaram negativas, e screening para imunodeficiência congênita, também sem alterações.
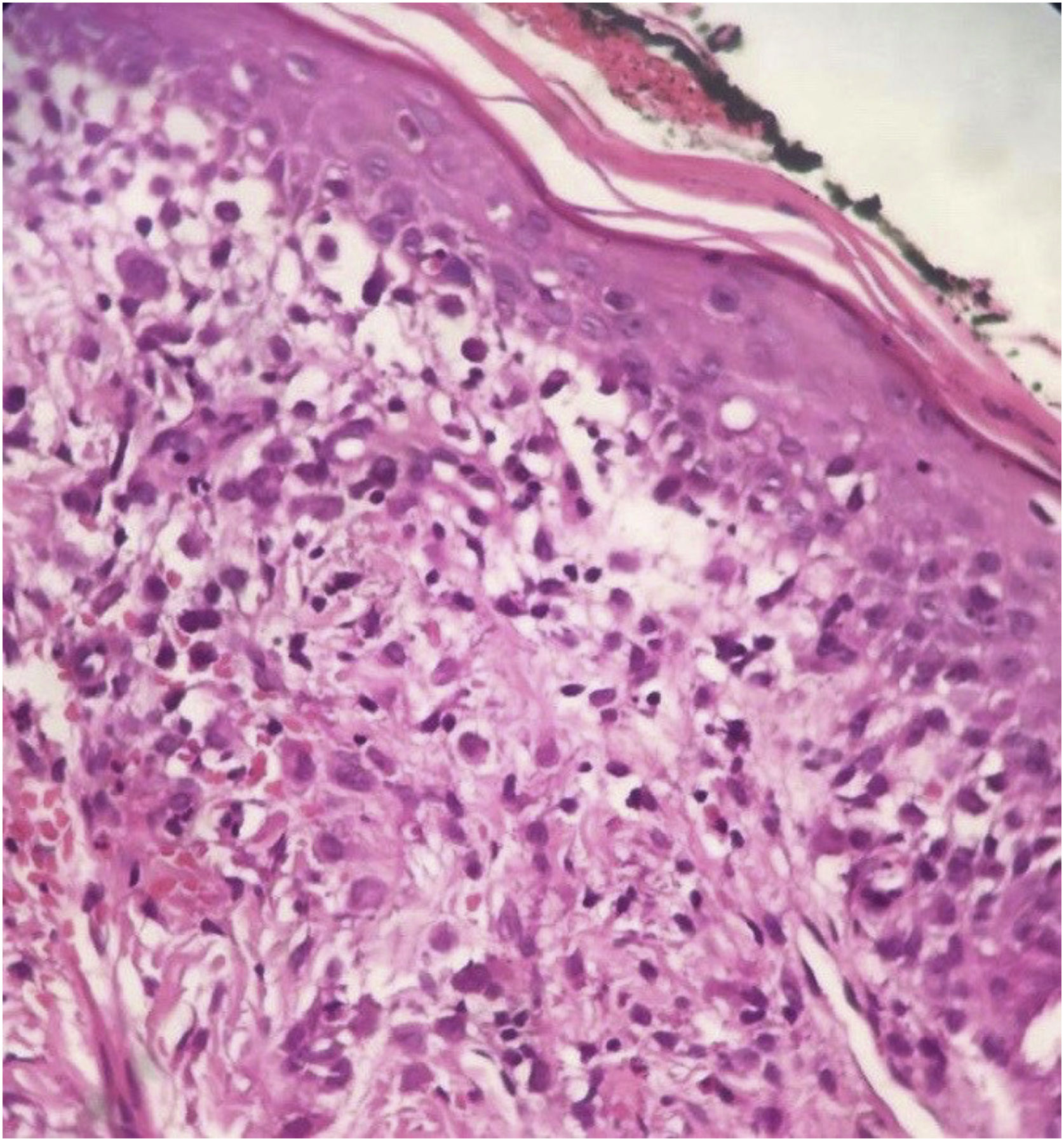
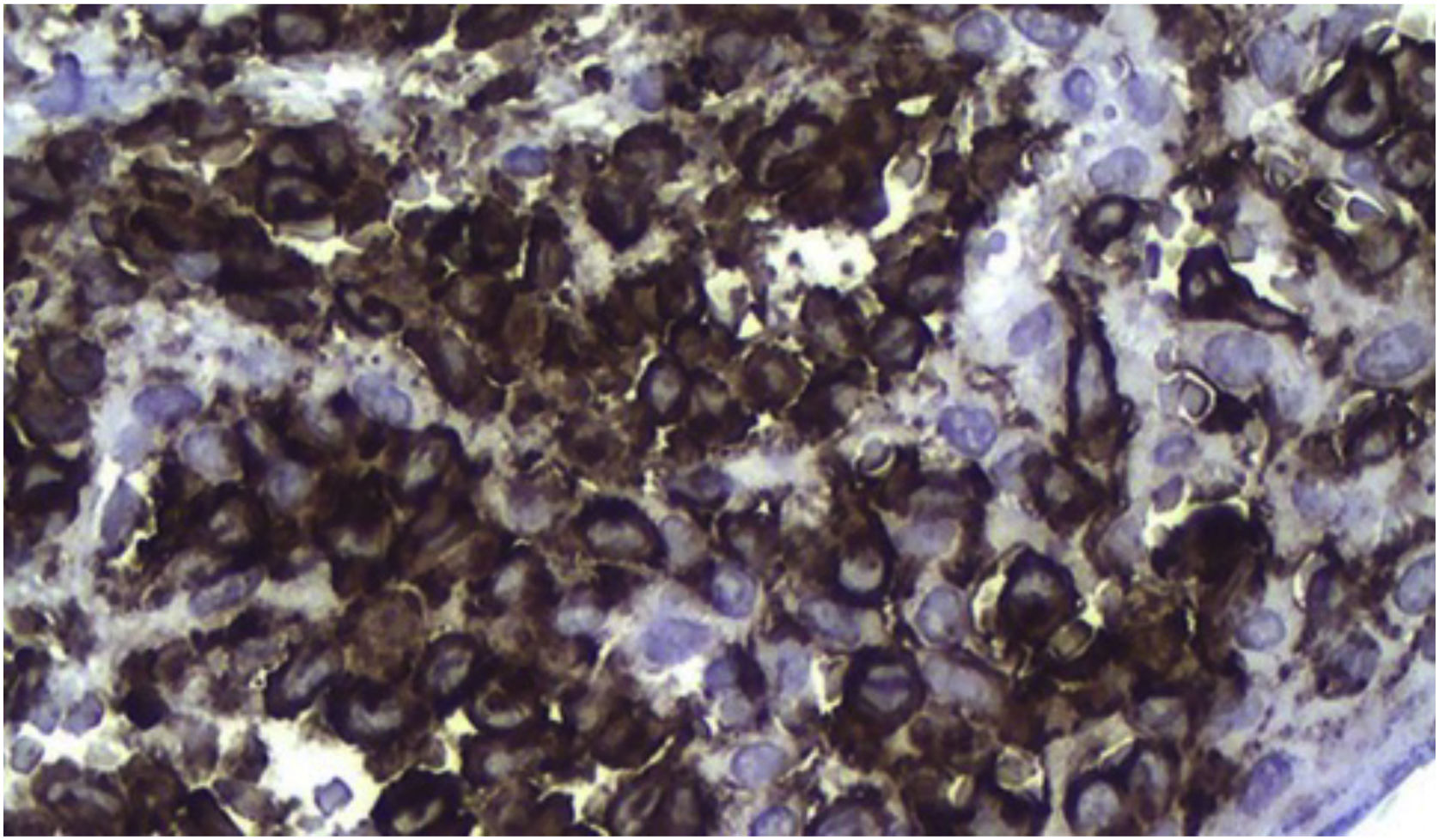
No anatomopatológico, achado de dermatite crônica associada à presença de células sugestivas de células de Langerhans (fig. 2). Na imuno‐histoquímica, positividade para CD1a, CD68, proteína S100; Ki67 positivo em 70% das células (fig. 3). Confirmado o diagnóstico de HCL, a investigação de outros órgãos por meio de mielograma e tomografias computadorizadas de tórax, abdome e pelve levou à classificação do caso como HCL forma multissistêmica em decorrência de acometimento pulmonar e hepático. Iniciado tratamento com vimblastina 3mg/m2 semanal+prednisona 20mg/m2, conforme Guideline da Sociedad Brasileira de Histyocite Society. O paciente apresentou melhora importante do quadro, mas após cerca de dois meses as lesões cutâneas retornaram e houve piora do quadro pulmonar, com necessidade de oxigenoterapia. Foi introduzido novo esquema quimioterapêutico com cladribina (2‐CdA), conforme protocolo japonês. O paciente segue estável, em acompanhamento conjunto com a Oncologia.
A incidência de HCL varia de dois a nove casos por milhão de crianças com menos de 15 anos; o pico é entre 1 e 3 anos de idade.2 Pode acometer um ou múltiplos órgãos; são considerados de risco: fígado, baço e medula óssea. A maioria dos pacientes apresenta envolvimento de sistema único (70%).1
O órgão mais frequentemente acometido é o osso, seguido pela pele, mas em lactentes as manifestações cutâneas são os principais achados. Dermatologicamente, apresentam‐se com quadro que lembram dermatite seborreica, e menos frequentemente como lesões hemorrágicas, apesar de essas direcionarem melhor para o diagnóstico.3
A patogênese é pouco esclarecida, com teorias que apoiam sua natureza reacional e neoplásica. A caracterização como desordem neoplásica é corroborada pelo encontro de mutação no gene BRAF V600E e ativação da via quinase MAP (MAPK).4
O diagnóstico depende da correlação clínica, anatomopatológica e imuno‐histoquímica. Na histologia, observam‐se proporções variadas de células de Langerhans com aspecto de “grão de café”. Imuno‐histoquímica positiva para CD1a e CD207 (langerina) fecham o diagnóstico.5
O tratamento será de acordo com a extensão e a gravidade da doença. Quando o acometimento é apenas cutâneo, a resolução espontânea é comum. Para doença multissistêmica, tratamento com esteroides sistêmicos e vimblastina por 12 meses é o regime de primeira linha.5
A HCL é doença grave que, por sua manifestação clínica muito diversa, é frequentemente diagnosticada tardiamente, atrasando o início do tratamento. A presença do dermatologista no corpo clínico possibilita seu reconhecimento precoce, com significante impacto no prognóstico dos pacientes.
Suporte financeiroNenhum.
Contribuição dos autoresThaís Oliveira Utiyama: Elaboração e redação do manuscrito; revisão crítica da literatura; revisão crítica do manuscrito.
Maria Laura Malzoni: Participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito; aprovação da versão final do manuscrito.
Thalita Gabrieli Sanches Vasques: Revisão crítica do manuscrito.
Cassiano Tamura Vieira Gomes: Aprovação da versão final do manuscrito.
Conflito de interessesNenhum.
Como citar este artigo: Utiyama TO, Malzoni ML, Vasques TG, Gomes CT. Langerhans cell histiocytosis: a rare case of the multisystemic form in an infant. An Bras Dermatol. 2023;98:398–9.
Trabalho realizado na Faculdade de Ciências Médicas e da Saúde de Sorocaba, Pontifícia Universidade Católica de São Paulo, Sorocaba, SP, Brasil.
