








Manifestações cutâneas ocorrem no curso das neoplasias hematológicas e precedem, acompanham ou são tardias em relação ao diagnóstico. Decorrem de fenômenos paraneoplásicos, das infiltrações tumorais, da imunossupressão decorrente da própria doença hematológica ou de seu tratamento. Cabe ao dermatologista ter conhecimento desses quadros que podem auxiliar tanto no diagnóstico da doença de base quanto na diminuição da morbidade dos doentes. Esta revisão (parte II) aborda as alterações dermatológicas paraneoplásicas associadas às neoplasias hematológicas sistêmicas.
Este artigo, parte II da revisão sobre as manifestações dermatológicas das neoplasias hematológicas, aborda as dermatoses inespecíficas (ou paraneoplásicas) das neoplasias hematológicas sistêmicas. As principais afecções dermatológicas desse contexto serão discutidas.
Define‐se dermatose paraneoplásica como um grupo de doenças cutâneas que apresentam forte associação com a presença de malignidade interna; elas precedem, acompanham ou são tardias ao aparecimento da neoplasia em questão, sem que exista a presença de células cancerígenas na pele.1,2 Algumas dermatoses ocorrem apenas na presença de uma neoplasia e são chamadas de dermatoses paraneoplásicas obrigatórias, enquanto outras ocorrem associadas a outras condições, e são denominadas dermatoses paraneoplásicas facultativas.2
A tabela 1 resume as manifestações dermatológicas paraneoplásicas, as neoplasias hematológicas associadas a essas e possíveis tratamentos. Na tabela 2 estão listadas as frequências dessas dermatoses conforme dados da literatura.
Manifestações cutâneas inespecíficas das neoplasias hematológicas: associações e manejo
| Diagnóstico Dermatológico | Alteração hematológica mais frequentemente encontrada | Manifestação dermatológica típica | Manejo |
|---|---|---|---|
| Síndrome de Sweet | LMA, neoplasias mieloides crônicas, LMMA | Nódulos, pápulas e placas, eritematosas ou violáceas e dolorosas | Corticoide sistêmico |
| Iodeto de potássio | |||
| Raramente: gamopatias monoclonais, MM, neoplasias linfoides | Dapsona | ||
| Colchicina | |||
| Pioderma gangrenoso | LMA | Forma clássica: úlcera dolorosa de bordas violáceas irregulares, subminadas, exsudato inflamatório estéril e base necrótica | Cuidados locais da ferida – óxido de zinco ou petrolatum nas bordas da ferida |
| Outras: SMD, LMC, PV, trombocitemia essencial, mielofibrose | |||
| Forma bolhosa: bolhas de borda azul acinzentada evoluindo para erosão/úlceras rasas – face e braços | Casos leves: Corticoide tópico de alta potência ou inibidores da calcineurina; Corticoides intralesionais | ||
| Forma vegetante: Lesões exofíticas eritematosas, e verrucosa – cabeça e pescoço | Casos extensos: Corticoide sistêmico; Ciclosporina | ||
| Forma pustulosa: pústulas dolorosas sobre base eritematosa | Outras: pulsoterapia com metilprednisolona, metotrexato, micofenolato, colchicina, sulfassalazina, dapsona, minociclina, apremilaste, talidomida | ||
| Imunobiológicos: infliximabe, adalimumabe, etanercepte, ustekinumabe | |||
| Doença refratária: imunoglobulina endovenosa, ciclofosfamida, clorambucil | |||
| Pustulose subcórnea | Paraproteinemia IgA | Pústulas com distribuição serpiginosa ‐ tronco e áreas intertriginosas | Dapsona |
| Outras: mieloma por IgA, anemia aplástica, linfomas, LMC, PV | Fototerapia, corticoides tópicos, corticoide sistêmico e acitretina | ||
| Pústulas maiores: secreção coletada na porção inferior (meio a meio). | |||
| Hidradenite écrina neutrofílica | LMA e LMC associados ou não a quimioterapia | Placas eritematoedematosas, pruriginosas ou dolorosas, dispersas ‐ mãos, face, extremidades | Corticoide sistêmico |
| Outras: LLC B, leucemia mielomonocítica, linfoma de ESL e não Hodgkin | Dapsona | ||
| Dermatose eosinofílica | LLC B | Pápulas, vesículas, bolhas ou nódulos pruriginosos. | Corticoide tópico |
| Outras: doenças linfoproliferativas de células B, linfomas T e leucemia agudas | Fototerapia | ||
| Semelhantes a reação a picada de inseto. | Corticoide sistêmico | ||
| Dapsona | |||
| Imunossupressores: metotrexato, azatioprina, lenalidomida, Dupilumabe | |||
| Prurido | Neoplasias mieloproliferativas (LMC, PV, mielofibrose primária), trombocitose essencial | Mais comum: prurido aquagênico – sensação de coceira, ardor, queimação ou pinicação depois do contato com a água na pele | Inibidores da recaptação de serotonina – paroxetina |
| Anticonvulsivantes ‐ gabapentina, pregabalina | |||
| Linfomas de Hodgkin e não Hodgkin | Antagonista dos receptores de opioides – Naltrexone | ||
| Fototerapia | |||
| Ácido acetilsalicílico na PV | |||
| Antagonista do receptor da neuroquina‐1: aprepitanto | |||
| Corticoides nos casos intratáveis | |||
| Talidomida nos pacientes paliativos | |||
| Vasculite cutânea de pequenos vasos | SMD | Púrpuras palpáveis dolorosas/pruriginosas | Tratamento da doença de base |
| Outras: LMA, LMC, mielofibrose, PV, trombocitemia essencial | |||
| Principalmente: membros inferiores distais | Corticoide sistêmico | ||
| Poliarterite nodosa | Leucemia de células pilosas | Nódulos subcutâneos, púrpura palpável, livedo reticular ou racemoso, ulcerações e bolhas | Corticoide sistêmico em altas doses |
| Principalmente: membros inferiores | Imunossupressores: metotrexato, ciclofosfamida | ||
| Eritema elevatum diutinum | Gamopatias monoclonais do tipo IgA | Pápulas, nódulos ou placas eritematovioláceas ‐ superfícies extensoras dos membros | Dapsona |
| Casos localizados: corticoide intralesional excisão cirúrgica | |||
| SMD | |||
| Pênfigo paraneoplásico | Linfoma não Hogdkin | Erosões progredindo para ulcerações orais e conjuntivais graves – toda superfície da orofaringe/vermelhão dos lábios | Prednisona |
| LLC | Ciclosporina | ||
| Doença de Castleman | Ciclofosfamida | ||
| Macroglobulinemia de Waldenstrom | Pseudoconjuntivite membranosa grave | Outros agentes: rituximabe, alemtuzumabe | |
| Ictiose adquirida | Linfoma de Hodgkin | Escamas romboides hipocrômicas/cinza, com aspereza/ descamação lamelar. | Hidratação cutânea |
| Outras: Linfoma não Hodgkin, linfoma cutâneo de células T, leiomiossarcoma, MF, MM | |||
| Agentes ceratolíticos | |||
| Semelhante a escama de peixe | |||
| Tronco e membros – superfícies extensoras, poupando flexuras | Tratamento da doença de base | ||
| Eritema Nodoso | LMA, LMC, LMMC | Nódulos sensíveis e eritematosos e placas de 1–6cm de diâmetro | Bandagens de compressão e elevação dos membros |
| Lesões simétricas em extremidades distais inferiores/pré‐tibiais | AINEs | ||
| Corticoide sistêmico, iodeto de potássio, colchicina, dapsona, hidroxicloroquina | |||
| Xantoma plano difuso | MM | Máculas ou placas amarelo‐alaranjadas irregulares, difusas, simétricas e assintomáticas | Remissão da condição hematológica resulta em melhora cutânea |
| Gamopatias monoclonais | |||
| Predileção por face, tronco e áreas intertriginosas | |||
| Escleromixedema | MM | Erupção generalizada de pápulas firmes, cerosas, de 2–3mm, cupuliformes/planas – mãos, antebraços, cabeça, pescoço, tronco e coxas superiores | Primeira linha: Imunoglobulina intravenosa |
| Outras: linfomas, macroglobulinemia de Waldenstrom e LMMA | Talidomida (ou lenalidomida) | ||
| Corticoide sistêmico | |||
| Distribuição linear com pele ao redor brilhante e endurecida (esclerodermoide) | Fotoquimioterapia extracorpórea | ||
| PUVA | |||
| Feixe de elétrons, corticoterapia/retinoides tópicos | |||
| Xantogranuloma necrobiótico | Gamopatia monoclonal e Mieloma Múltiplos | Múltiplas pápulas e nódulos endurecidos, assintomáticos, amarelados a marrom‐avermelhados. | Corticoides tópicos e sistêmicos, talidomida, imunoglobulina intravenosa de alta dose (IVIG), clorambucil, dentre outros |
| Outras: Linfoma não Hodgkin e de Hodgkin, macroglobulinemia de Waldenstrom, SMD, LLC | |||
| Evolução lenta para grandes placas. | |||
| Prurigo | Leucemia e doença de Hodgkin | Pápulas simetricamente distribuídas, nódulos hiperceratóticos ou escoriados e cicatrizes. | Diagnóstico e tratamento da doença de base |
| Membros e tronco – áreas acessíveis a coçadura | Corticoide tópico, fototerapia e anti‐histamínicos – pouco efetivos | ||
Dados epidemiológicos das manifestações cutâneas inespecíficas das neoplasias hematológicas
| Manifestação | Associação com doenças hematológicas |
|---|---|
| Síndrome de Sweet | 20% relacionados a malignidades hematológicas |
| 55% na forma histiocítica | |
| Pioderma gangrenoso | 3,9‐7% relacionados a malignidades hematológicas |
| Pustulose subcórnea | Prevalência desconhecida |
| Hidradenite écrina neutrofílica | Prevalência desconhecida |
| 67% associados a LMA que receberam quimioterapia | |
| Dermatose eosinofílica | Dermatose paraneoplásica associada principalmente a LLC B |
| Prurido | 2% relacionados a malignidades hematológicas |
| Vasculite cutânea de pequenos vasos | 3,8–8% relacionados a malignidades hematológicas |
| Poliarterite nodosa | Prevalência desconhecida |
| Eritema elevatum diutinum | Prevalência desconhecida |
| 16%–42% – anormalidade hematológica de base | |
| Pênfigo paraneoplásico | 84% relacionados a malignidades hematológicas |
| Ictiose adquirida | Prevalência desconhecida |
| Eritema nodoso | 0%–4% relacionados a malignidades hematológicas em revisões de casos da literatura |
| Xantoma plano difuso | 48% em revisões de casos da literatura |
| Escleromixedema | Gamopatia monoclonal é um dos critérios diagnósticos, sendo atípico não ser encontrada |
| Xantogranuloma necrobiótico | 80% dos casos: gamopatia monoclonal associada |
| 10% evoluem para MM |
As dermatoses neutrofílicas são um grupo de manifestações cutâneas caracterizadas por lesões polimórficas, acompanhadas no exame histopatológico por infiltrado dérmico de polimorfonucleares.3 Incluem a síndrome de Sweet, o pioderma gangrenoso, a pustulose subcórnea e a hidradenite écrina neutrofílica.3,4 Lesões de transição ou overlap entre as formas clínicas também foram descritas, assim como comprometimento extracutâneo.5
Os mecanismos fisiopatológicos implicados em seu desenvolvimento incluem aumento de expressão de citocinas pró‐inflamatórias responsáveis pelo recrutamento e migração de neutrófilos para a pele, como IL‐1, IL‐8, IL‐17 e TNF‐alfa.6 Na patogênese, fatores genéticos também são implicados: pacientes com dermatoses neutrofílicas apresentam mutações que ocorrem também em doenças autoinflamatórias, além de mutações em genes reguladores da imunidade inata.6
Três formas clínico‐patológicas de dermatoses neutrofílicas foram descritas: forma profunda ou hipodérmica, que inclui o pioderma gangrenoso; forma em placas ou dérmica, que inclui a síndrome de Sweet, e forma superficial ou epidérmica, que inclui a pustulose subcórnea.7
As dermatoses neutrofílicas estão classicamente associadas a malignidades hematológicas e ocorrem de modo concomitante ao diagnóstico, antes ou depois. São mais frequentemente associadas às malignidades mieloides, como leucemia mieloide aguda (LMA), neoplasias mieloides crônicas (LMC), leucemia mielomonocítica (LMMC) e mais raramente associadas às gamopatias monoclonais, mieloma múltiplo (MM) e neoplasias linfoides.8 Especificamente no caso de neoplasias mieloides, há evidência de que os neutrófilos que infiltram a pele são clones relacionados às células neoplásicas.9
Alguns fármacos utilizados no tratamento de neoplasias hematológicas podem induzir o desenvolvimento de dermatoses neutrofílicas, como azacitidina, imatinibe, lenalidomida e fator estimulador de colônia de granulócitos.6 Dermatoses neutrofílicas também podem ocorrer como evento adverso cutâneo a inibidores de checkpoint.10
Síndrome de SweetA síndrome de Sweet ou dermatose neutrofílica aguda febril é síndrome inflamatória caracterizada pelo início abrupto de nódulos, pápulas e placas (fig. 1), eritematosas ou violáceas, dolorosas, acompanhadas por febre e leucocitose.3
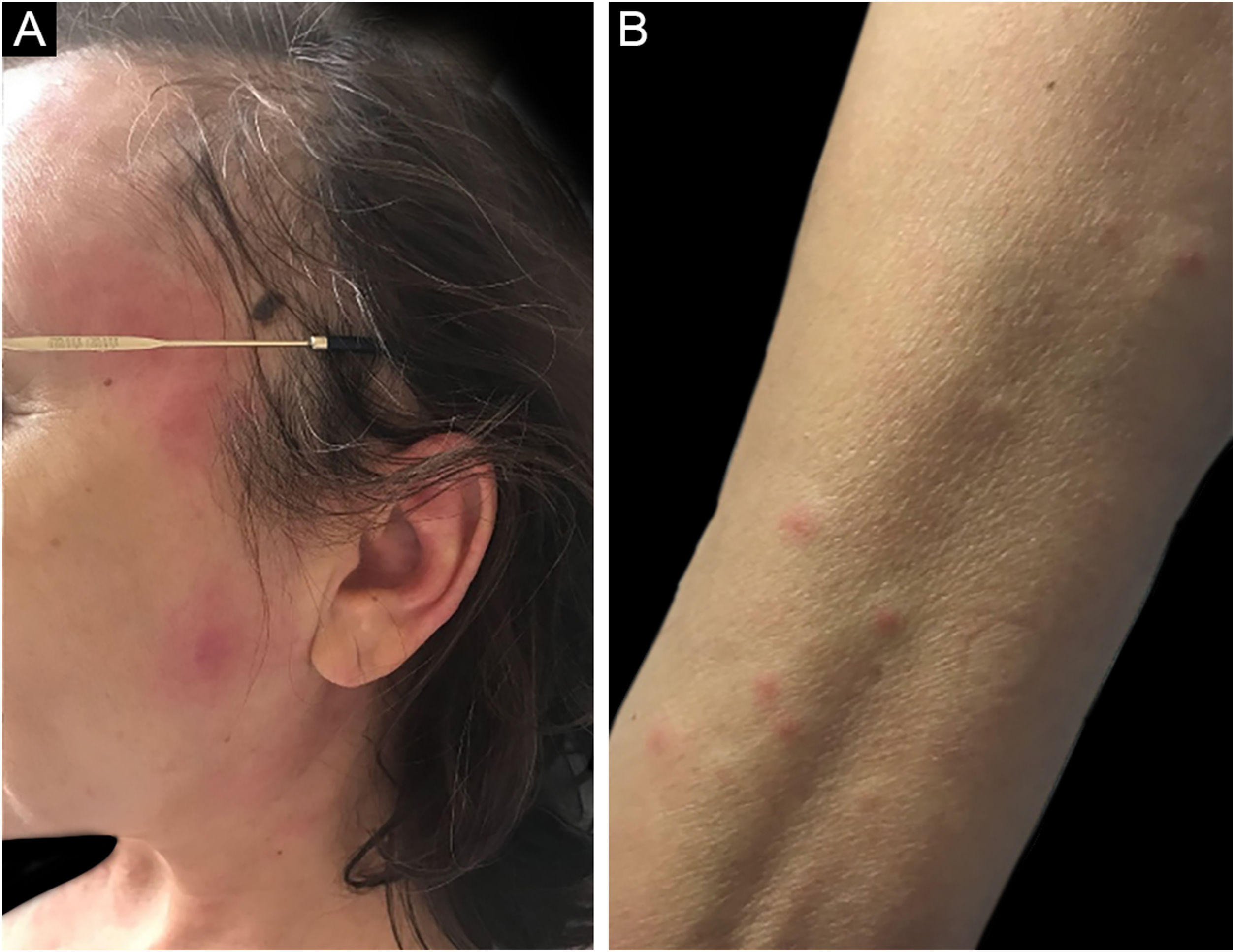
Formas clínicas subcutâneas semelhantes a celulite extensa já foram descritas e estão frequentemente associadas a malignidades hematológicas, enquanto formas bolhosas e ulceradas ocorrem também na síndrome de Sweet idiopática.6 Achados extracutâneos incluem artralgia ou artrite, envolvimento ocular, alveolite, hepatite, miosite, meningite asséptica e envolvimento gastrintestinal.4 Fenômeno de patergia pode ocorrer, e 20% dos pacientes têm prurido associado.11 É recorrente em aproximadamente 1/3 dos casos.7
Três variantes foram descritas: idiopática ou clássica, relacionada a fármacos e associada a malignidades. A síndrome de Sweet associada a malignidades corresponde a 20% do total de casos.12
Sua patogênese envolve reação de hipersensibilidade a antígenos tumorais ou alterações no perfil de citocinas, com altos níveis de IL‐4, IL‐6, interferon‐gama e fator estimulador de colônia de granulócitos ou fatores de suscetibilidade genética.6
O diagnóstico depende da presença dos critérios clínicos e exame histopatológico, que demonstra infiltrado neutrofílico dérmico sem vasculite. Pode haver também leucocitoclasia e edema endotelial.13
Uma variante infrequente da síndrome de Sweet, também associada a malignidades hematológicas, é a síndrome de Sweet histiocítica. Tem aspecto clínico semelhante à síndrome de Sweet clássica, mas apresenta infiltrado dérmico de células mielomonocíticas com morfologia histiocitoide.14 Até 55% dos casos de síndrome de Sweet histiocítica são associados a malignidades, entre as quais se destaca a síndrome mielodisplásica.15
O tratamento é tipicamente realizado com corticoide sistêmico, na dose de 0,5 a 1 mg/kg de prednisona. Alternativas para casos com contraindicação para uso de corticoide incluem iodeto de potássio (300mg, 3 vezes/dia); dapsona, com dose alvo de 100 a 200 mg/dia e colchicina, na dose de 1 a 1,5 mg/dia.16
Pioderma gangrenosoPioderma gangrenoso é dermatose neutrofílica incomum, que se inicia por pápula ou pústula dolorosa que evolui para nódulo e, posteriormente, evolui para ulceração.3 É tipicamente diagnóstico de exclusão, após afastadas outras causas infecciosas ou vasculares de úlceras.17
Na patogênese do pioderma gangrenoso, aumento de citocinas e fatores quimiotáticos de neutrófilos, como IL‐1β, IL‐17, TNF‐α, IL‐8, IL‐6, IL‐17 e IL‐23, já foi demonstrado. Além disso, há aumento de expressão de metaloproteinases (MMP), em especial MMP 9 e 10.18
O quadro clínico, na forma clássica, caracteriza‐se por úlcera com bordas violáceas irregulares, subminadas, exsudato inflamatório estéril e base necrótica. As lesões são tipicamente dolorosas, e a dor é mais intensa nos períodos de progressão das lesões.7 O fenômeno de patergia foi descrito em 31% dos casos,19 e em pacientes com história prévia de pioderma gangrenoso submetidos a procedimento cirúrgico houve desencadeamento de lesões em até 15% dos casos.20 Após a resolução do quadro, há formação de cicatrizes cribiformes.6
Foram descritas três outras formas clínicas: bolhosa, pustulosa e vegetante ou granulomatosa. As três formas têm em comum com a forma clássica o início da lesão como pápula ou pústula que evolui para nódulo. Na forma bolhosa (fig. 2), as lesões ocorrem nos braços ou na face e caracterizam‐se por bolhas de borda azul acinzentada que posteriormente evoluem para erosão e formação de úlceras rasas. A forma pustulosa apresenta‐se com pústulas dolorosas sobre base eritematosa e pode ser acompanhada de febre e artralgias. A forma vegetante ou granulomatosa ocorre preferencialmente na cabeça e pescoço e caracteriza‐se por úlceras que gradualmente se transformam em lesões exofíticas eritematosas, de superfície verrucosa.7,17 A forma periostomal ocorre em associação a doenças inflamatórias intestinais e pode ser exacerbada por fenômeno de patergia.21
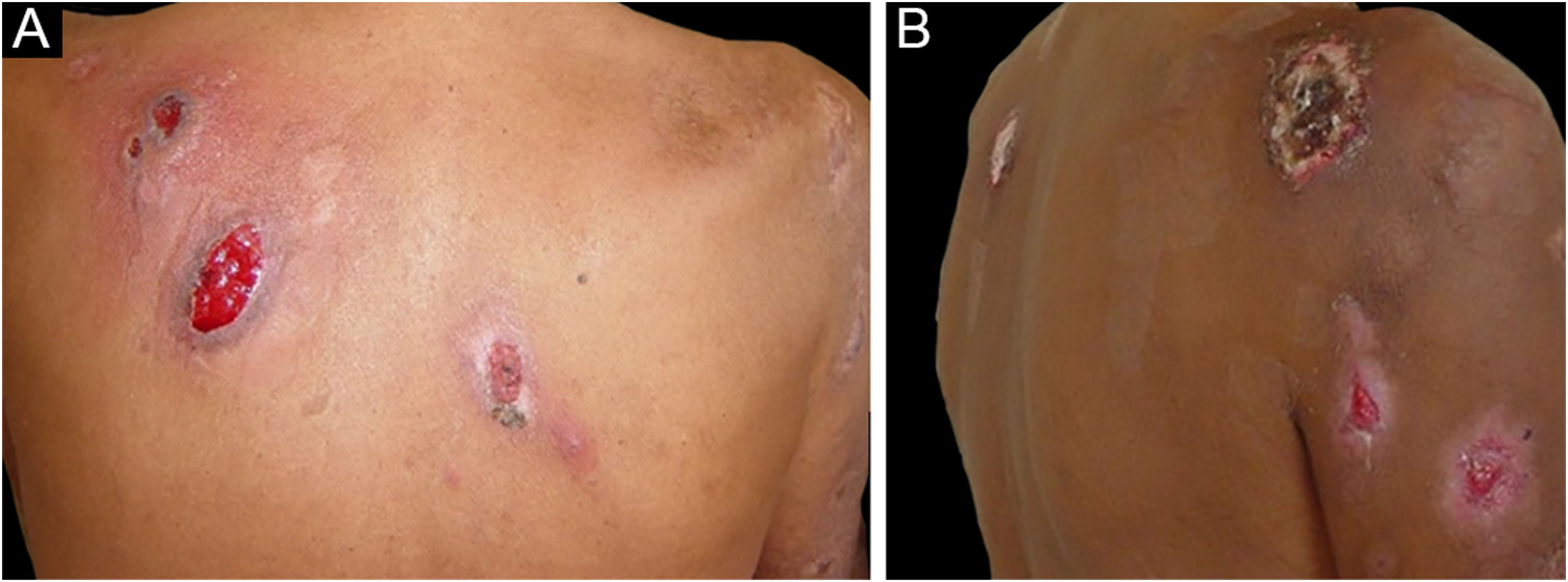
Associação do pioderma gangrenoso com doenças sistêmicas ocorre em 50% dos casos.3 As principais afecções relacionadas são: colite ulcerativa, doença de Crohn, artrite e malignidades hematológicas, estas últimas presentes em 3,9% a 7% dos doentes. Entre as malignidades hematológicas, MA, LMC, MM e gamopatia monoclonal assintomática de significado incerto (MGUS) estão mais associadas.3,6,22 A forma bolhosa, que corresponde a 50% dos casos de pioderma gangrenoso observado no câncer hematológico, frequentemente é observada na LMA, mas ocorre também na síndrome mielodisplásica (SMD), LMC, policitemia vera (PV), trombocitemia essencial e mielofibrose.4 A forma pustulosa é mais associada a doença inflamatória intestinal.17 O desenvolvimento do pioderma gangrenoso nos casos de neoplasia hematológica confere pior prognóstico à doença de base.23
Para o diagnóstico histopatológico, é preferível realização de excisão fusiforme que inclua a borda e o fundo da úlcera.7 No exame histopatológico, há infiltrado supurativo de neutrófilos na derme e subcutâneo, acompanhado por hemorragia e necrose. Para o diagnóstico é necessário excluir causas infecciosas, com coloração para fungos e micobactérias.7
O tratamento sempre envolve cuidados locais, com limpeza e curativos apropriados para cada caso. É importante o cuidado com as bordas das lesões, onde pode ser utilizado óxido de zinco ou petrolatum.23 Também é primordial a analgesia adequada.18
Nos casos leves, pode ser realizado tratamento tópico, com corticoide de alta potência como clobetasol, inibidor de calcineurina ou corticoide intralesional.24 Para casos extensos, está indicado tratamento com corticoide sistêmico (0,5 a 1 mg/kg/dia) e, após a melhora, seu desmame deve ser lento para evitar recidivas. A ciclosporina na dose de 4 mg/kg/dia tem resposta rápida e eficácia comparável ao corticoide, mas seu uso nos casos associados a malignidades hematológicas é controverso.6,18 Outras opções sistêmicas incluem pulsoterapia com metilprednisolona, metotrexato, micofenolato, colchicina, sulfassalazina, dapsona, minociclina, apremilaste e talidomida.18
Imunobiológicos também são usados no tratamento do pioderma gangrenoso. A introdução do imunobiológico deve ser considerada opção precoce. Em ensaio clínico randomizado, 69% dos pacientes tratados com infliximabe apresentaram melhora e 21% apresentaram remissão seis semanas após infusão da primeira dose.25 Além do infliximabe, também são utilizados adalimumabe, etanercepte, ustekinumabe.18 Na doença refratária às demais alternativas, consideram‐se imunoglobulina endovenosa, ciclofosfamida ou clorambucil.
Pustulose subcórneaA pustulose subcórnea é dermatose rara caracterizada por pústulas subcórneas flácidas, de curso crônico.
Em sua patogênese, nas camadas mais superficiais da epiderme, identifica‐se presença de citocinas relacionadas à migração de neutrófilos, como TNF‐alfa, IL‐8, C5a.26
As lesões usualmente ocorrem no tronco e em áreas intertriginosas e distribuem‐se de modo serpiginoso ou anular. As pústulas em geral são pequenas e aparecem em surtos, sobre pele normal ou discretamente eritematosa. Nas pústulas maiores, a secreção fica coletada na porção inferior da lesão, aspecto conhecido como pústula meio a meio. Na evolução, há possibilidade de discreta hiperpigmentação após a resolução das pústulas.26
A pustulose subcórnea associa‐se a doenças hematológicas, em especial a paraproteinemia por IgA. Outras associações incluem mieloma por IgA, anemia aplástica, linfomas, LMC, policitemia vera.6
No exame histopatológico, há presença de abscesso neutrofílico subcórneo, com ou sem infiltrado perivascular superficial misto e ausência de espongiose.3
O tratamento é realizado com dapsona, iniciada na dose de 25mg, com alvo de 50 a 150mg/dia. A menor dose necessária para controle dos sintomas deve ser mantida, e monitoramento de toxicidade hematológica é mandatório.26 Outras opções terapêuticas incluem fototerapia, corticoides tópicos, corticoides orais e acitretina.6
Hidradenite écrina neutrofílicaA hidradenite écrina neutrofílica é dermatose neutrofílica incomum originalmente descrita em pacientes com LMA tratados com citarabina e posteriormente identificada em casos não tratados de LMA e LMC. Nos casos associados à quimioterapia, foi considerada toxicidade direta do fármaco à glândula sudorípara écrina.4 Em revisão de literatura com 51 casos de hidradenite neutrofílica écrina, 67% eram de LMA que haviam recebido quimioterapia.27 Outras condições associadas são leucemia linfocítica crônica (LLC) B, LMMC, linfoma de Hodgkin (LH) e linfoma não Hodgkin (LNH).6
O quadro clínico é caracterizado por placas eritematoedematosas que podem ser pruriginosas ou dolorosas, dispersas nas mãos, face, extremidades (fig. 3). Existe a possibilidade de haver febre, principalmente nos casos com neutropenia associada.6
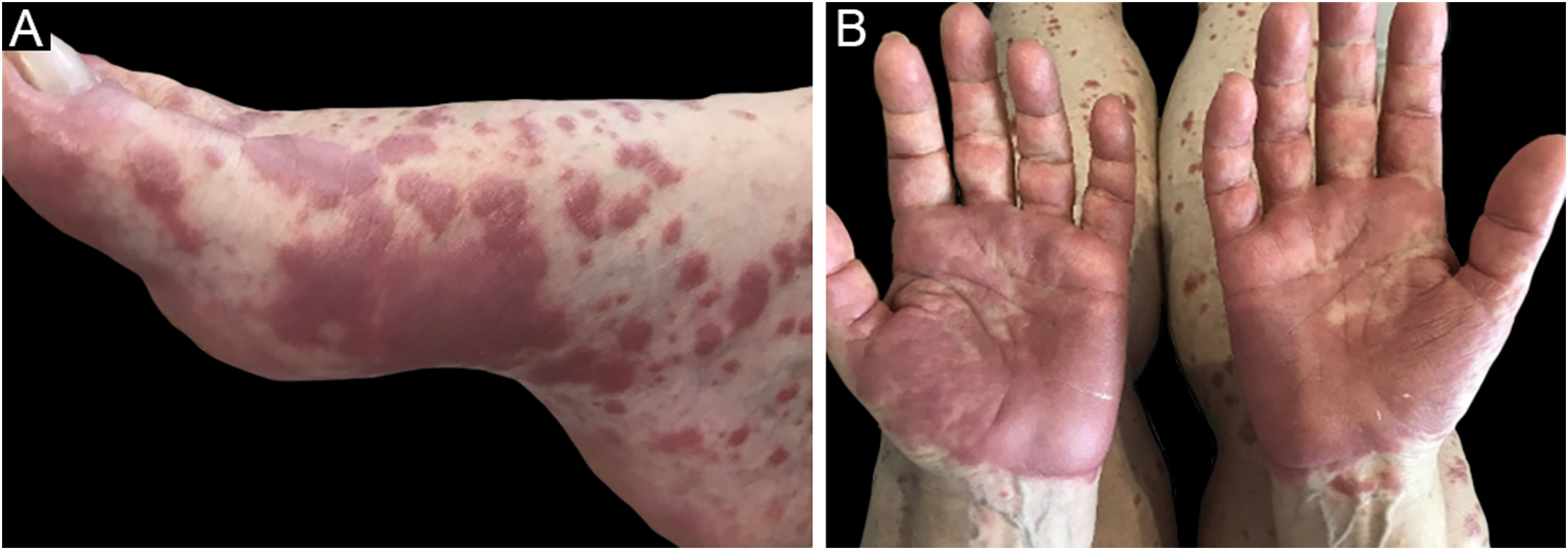
O diagnóstico histopatológico envolve infiltrado neutrofílico nas glândulas sudoríparas écrinas e nos ductos écrinos. Pode haver formação de abscesso intraductal, metaplasia siringoescamosa das glândulas sudoríparas e fibrose da derme adjacente.27
Alguns casos de hidradenite écrina neutrofílica têm resolução espontânea; porém, na experiência dos autores, a qualidade de vida do paciente é muito impactada e o manejo pode ser desafiador. Vários tratamentos são preconizados, como corticoide tópico ou sistêmico e dapsona.4
Dermatoses eosinofílicasAs dermatoses eosinofílicas fazem parte do grupo de dermatoses paraneoplásicas nos pacientes com malignidades hematológicas e se caracterizam clinicamente por pápulas, vesículas, bolhas ou nódulos pruriginosos, possivelmente dolorosos, semelhantes à reação a picada de inseto, dispersos por todo o corpo. As lesões podem se desenvolver sobre base eritematoedematosa, urticariforme.28
As dermatoses eosinofílicas estão associadas principalmente à LLC B e outras doenças linfoproliferativas B, como linfoma do manto, leucemia linfoblástica aguda, linfoma de grandes células B, mas também com linfomas T e leucemias agudas.6,28
A patogênese inclui aumento de produção de IL‐4 e IL5 desencadeado por picada de inseto, exposição a fármacos ou vírus, que leva a alteração da resposta imune com predominância de proliferação de eosinófilos.29 Outra possibilidade é que a doença hematológica ou seu tratamento provoque predominância de repertório T‐helper 2 (Th2), que favoreceria recrutamento de células T para a pele e ativação de eosinófilos em resposta a antígenos ambientais.6
Três formas clínicas foram descritas: bolhosa, caracterizada por vesicobolhas semelhantes ao penfigoide bolhoso (fig. 4); tipo reação a picada de inseto, caracterizada por pápulas e placas urticariformes; e celulite‐símile, caracterizada por placas e nódulos semelhantes à síndrome de Wells.6 As lesões, em geral, ocorrem meses ou anos depois do diagnóstico hematológico, mas em raros casos precedem o quadro hematológico.30
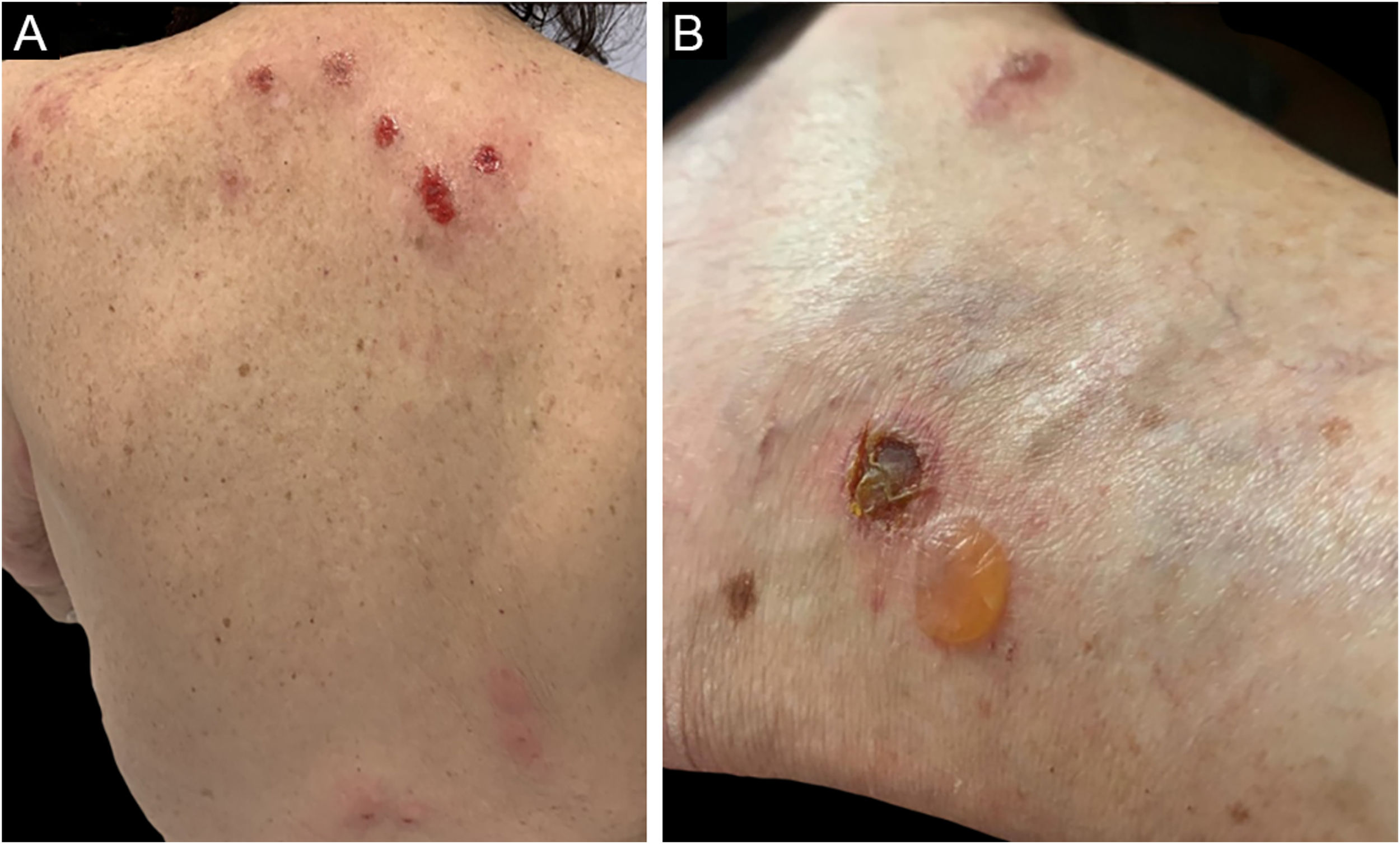
Os seguintes critérios são utilizados no diagnóstico: 1) pápulas, nódulos ou vesicobolhas pruriginosos refratários aos tratamentos convencionais; 2) presença de infiltrado linfo‐histiocítico superficial e profundo, rico em eosinófilos; 3) exclusão de outras causas de eosinofilia tecidual; 4) diagnóstico de malignidade hematológica.31
O exame histopatológico demonstra infiltrado inflamatório linfocitário dérmico perivascular e perianexial rico em eosinófilos, que pode se estender até o subcutâneo. Figuras em chama de vela podem estar presentes e são encontradas também na síndrome de Wells, celulite eosinofílica, penfigoide bolhoso, farmacodermias e outras doenças com acúmulo de eosinófilos na derme.32
O tratamento das dermatoses eosinofílicas inclui corticoide tópico, fototerapia, corticoide sistêmico, dapsona, imunossupressores como metotrexato e azatioprina, lenalidomida e foi relatado tratamento com dupilumabe. O tratamento concomitante da doença hematológica de base pode contribuir para a melhora do quadro.28,33
PruridoPrurido é definido como sensação não agradável que provoca o desejo de coçar. É considerado um dos sintomas que mais incomodam e alteram a qualidade de vida do doente, até mesmo debilitante em muitos casos.34–36 Pode ser o primeiro sintoma de envolvimento sistêmico hematológico e precede o diagnóstico da doença de base em mais de 50% dos casos, com latência de meses a até 38 anos.34,36,37
A patogênese do prurido é complexa; estudos sugerem a ativação de uma via distinta de fibras C não mielinizadas via trato espinotalâmico a várias partes do cérebro. Essa via é influenciada por células inflamatórias variadas por meio de complexas interações com a liberação de citocinas, proteases e neuropeptídios.35,37
As neoplasias mieloproliferativas como LMC, policitemia vera, mielofibrose primária e trombocitemia essencial têm o prurido como o segundo sintoma mais referido pelo paciente em 56% dos casos, atrás somente da fadiga. Os linfomas tanto Hodgkin quanto não Hodgkin também são associados a prurido, assim como as SMDs primárias.34 Apesar disso, as causas hematológicas são relacionadas em apenas 2% do total de casos.35,37
O prurido é encontrado em até 69% dos doentes com PV segundo diferentes estudos.34,38 Usualmente, ele aparece como prurido aquagênico, isto é, sensação de coceira, ardor, queimação ou pinicação depois do contato com a água na pele, que dura poucos minutos, sem formar qualquer lesão.38 Cerca de 25% dos pacientes referem quadro debilitante que atrapalha as atividades diárias.34,36 A patogênese do prurido aquagênico na PV envolve o aumento de células mononucleares e eosinófilos na papila dérmica, edema, vasodilatação, aumento da atividade fibrinolítica e ativação da acetilcolinesterase. A mutação do JAK2V617F, encontrada na PV, promove ativação e hipersensibilidade dos basófilos.34
A mielofibrose primária é doença caracterizada pela mieloproliferação, fibrose reativa da medula óssea, osteoesclerose, angiogênese, hematopoiese extramedular e alteração na expressão das citocinas. O prurido é descrito em até 16% dos casos e foi associado em um estudo à leucocitose. Do total de pacientes com prurido relacionado à mielofibrose, cerca da metade tem prurido aquagênico.34,37,39,40
No LH, o prurido está presente em quase 40% dos pacientes e é severo em cerca de 6%. Pode preceder a doença em até quatro anos e normalmente ocorre nas pernas e à noite. Também é descrita hiperidrose transitória antes do aparecimento do prurido entre os dedos e nas palmas. A patogênese é ligada a resposta Th2, produção de IL‐10, IL‐4, IL‐5, IgE e hipereosinofilia. Já no LNH, o prurido é achado raro, exceto nos linfomas T cutâneos, que não serão abordados nesta revisão.
Na suspeita do prurido causado por doença neoplásica hematológica, a investigação inicial deve incluir a história detalhada e exame físico geral. Os exames iniciais de screening citados são hemograma completo, VHS, DHL, RX de tórax e US de abdome. Na suspeita de PV, a pesquisa da mutação do JAK2V617 é fundamental.35 De acordo com os resultados da avaliação inicial, a investigação deve ser ampliada conjuntamente com o hematologista para a realização de biópsia de medula óssea e exames de imagens específicos e direcionados a cada caso.
O tratamento do prurido ligado a doenças hematológicas é um desafio. O tratamento de escolha voltado para a doença de base é sempre a melhor solução, apesar de a eficácia não ser diretamente proporcional. Questiona‐se que isso ocorra porque a causa do sintoma não é diretamente ligada à presença das células anômalas, mas sim à reação imunológica que acompanha a doença.35,37 Medidas sintomáticas para controle do prurido são necessárias em muitos casos pelo importante decréscimo da qualidade de vida. Os anti‐histamínicos, muito prescritos, não aliviam o sintoma na maioria dos doentes e, dentre esses, os de primeira geração levam a melhores resultados quando comparados ao de segunda geração, pela sedação que causam. Os inibidores da recaptação da serotonina, como a paroxetina, os anticonvulsivantes gabapentina e pregabalina, e antagonistas dos receptores opioides como naltrexone são relativamente eficazes no controle desse sintoma. Fototerapia também é eficiente em muitos casos.34,35,37,39 A aspirina demonstra moderados resultados em casos de PV, e estudos recentes são animadores com uso de inibidores da JAK e mTOR no prurido da PV e mielofibrose primária.40 Corticoides podem ser usados no controle do prurido dos linfomas intratáveis.35 Recentemente, o aprepitanto, antagonista do receptor da neuroquina‐1, que já é usado nos pruridos dos linfomas cutâneos e dos tumores sólidos, tem sido citado no prurido das doenças hematológicas, ainda em poucos casos, com bons resultados.41 O manejo do prurido em pacientes paliativos pode exigir outras opções, como a talidomida, que é evitada no uso convencional a pacientes oncológicos pela gama de efeitos colaterais, principalmente a neuropatia periférica, que se sobrepõe a outros fármacos já em uso.35
PrurigoO prurigo crônico (PC) é caracterizado por intenso prurido associado a numerosas pápulas simetricamente distribuídas, nódulos hiperceratóticos ou escoriados e cicatrizes, principalmente, nos membros inferiores e tronco e em áreas acessíveis a coçadura.42,43 A pele entre as lesões é preservada.44
O PC como sinal de doença paraneoplásica é visto na leucemia e doença de Hodgkin,43 e é raro no LNH.45
Os pacientes que apresentam PC devem ter história clínica minuciosamente realizada; recomenda‐se exame físico cuidadoso, incluindo a palpação dos gânglios linfáticos, fígado e baço.46 Exames complementares devem ser realizados para descartar comorbidades e/ou malignidades subjacentes, de acordo com os achados de anamnese e exame físico.42 Sugere‐se acompanhamento clínico frequente, com repetição dessa investigação a cada três a seis meses para o diagnóstico definitivo precoce de possível malignidade subjacente.46
O tratamento inicial do PC paraneoplásico com corticoide tópico, fototerapia e anti‐histamínicos mostrou‐se pouco efetivo.45 O diagnóstico e tratamento da doença de base induz melhora significativa da doença, com controle das lesões cutâneas e do prurido.45
VasculitesO termo vasculite cutâneo inclui grande variedade de entidades clínicas que apresentam como característica comum achados histopatológicos com inflamação perivascular e dano a vasos sanguíneos. Não há consenso sobre a melhor forma de dividir as vasculites. Nesta revisão, abordaremos como neoplasias hematológicas relacionam‐se à vasculite cutânea de pequenos vasos (VCPV). Também abordaremos outras duas vasculites com achados cutâneos de características clínicas ou laboratoriais distintas: poliarterite nodosa (PAN), eritema elevatum diutinum (EED).
Vasculite cutânea de pequenos vasosA VCPV tem múltiplas etiologias, e é associada a neoplasias em 3,8%‐8% dos casos, principalmente doenças linfoproliferativas.3,4,6 As desordens hematológicas mais comumente encontradas são as SMDs, porém também há relatos de LMA, LMC, mielofibrose, PV e trombocitemia essencial.3,4,6,47 Pacientes com vasculites paraneoplásicas costumam ser mais velhos que pacientes com outras vasculites e tipicamente não se encontram na história fatores precipitantes clássicos como infecções ou uso de medicações.48,49
O mecanismo pelo qual as vasculites paraneoplásicas ocorrem não é plenamente elucidado, porém acredita‐se que uma conjunção de fatores seja responsável: clearance anormal de imunocomplexos, ligação de anticorpos não específicos nas paredes vasculares e produção desregulada de imunoglobulinas.48 Nos casos hematológicos, sugere‐se que o aumento da viscosidade sanguínea contribua para a redução do clearance de imunocomplexos nos pequenos vasos da derme.6
A apresentação clínica mais comum são púrpuras palpáveis que não desaparecem à digitopressão, geralmente nos membros inferiores distais, porém também acometendo membros superiores e tronco. As lesões são dolorosas ou pruriginosas.48,50 Raramente as lesões coalescem em placas ou formam bolhas hemorrágicas.4 As vasculites associadas às neoplasias hematológicas apresentam duração maior do que as não neoplásicas.6 Sintomas constitucionais podem estar presentes, como perda de peso e anorexia, artrite e artralgia, polineuropatia e dor abdominal, e são mais comuns que em pacientes com vasculite sem neoplasia.48 Na maioria dos casos, a vasculite ocorre simultaneamente ao diagnóstico da neoplasia subjacente; é possível preceder o diagnóstico ou mesmo ocorrer tardiamente.51
Achados histopatológicos incluem infiltração neutrofílica perivascular, necrose fibrinoide das paredes vasculares, presença de poeira nuclear e eritrodiapedese.4,6,48 O exame anatomopatológico é indistinguível de vasculites não paraneoplásicas.49 Entretanto, o achado de células neoplásicas no infiltrado inflamatório sugere o diagnóstico de vasculite leucêmica, apresentação rara da leucemia cútis associada a pior prognóstico.6 Pode‐se encontrar fator reumatoide e crioglobulinas positivas, geralmente em títulos baixos e não significativas para diagnóstico de outras entidades.48
O tratamento da vasculite é dependente do tratamento da neoplasia associada, assim como seu prognóstico.48 Em casos refratários, corticoterapia sistêmica pode ser empregada; os casos paraneoplásicos são mais resistentes ao tratamento.4,6 A melhora da vasculite com a quimioterapia para tratamento da desordem hematológica com posterior retorno das lesões cutâneas é sinal de recidiva neoplásica.6
Poliarterite nodosaA PAN é vasculite com acometimento de vasos de pequeno e médio calibre de múltiplos órgãos, incluindo a pele. Dentre as neoplasias hematológicas, a PAN está mais associada a leucemia de células pilosas.4 Dentre as malignidades mieloides, as patologias mais encontradas são a SMD e a LMMC.47
O rim é o órgão mais afetado na PAN. Comprometimento renal ocorre em 70%–80% dos pacientes e manifesta‐se com insuficiência renal, hipertensão, proteinúria ou mesmo hematúria. Hemorragias perirrenais espontâneas e bilaterais parecem ser mais comuns em pacientes com SMD, assim como hemorragias em outros órgãos. Esse fato é provavelmente causado pela presença simultânea de trombocitopenia e outras desordens da coagulação em combinação com anormalidades vasculares.47
Na pele, a PAN paraneoplásica apresenta‐se igual à forma clássica: nódulos subcutâneos, púrpura palpável, livedo reticular ou racemoso, ulcerações e bolhas. Os membros inferiores são preferencialmente acometidos, seguidos dos membros superiores e tronco. Outras manifestações sistêmicas incluem febre, poliartrite assimétrica, dor abdominal e neuropatias periféricas.3,4
O achado histopatológico típico é de vasculite necrotizante de vasos de médio calibre encontrado na pele ou em outro órgão acometido. P‐ANCA é encontrado em até 20% dos casos de PAN cutânea, geralmente ausente na vasculite sistêmica.4
O tratamento é feito com base no controle da doença subjacente, corticoterapia sistêmica em altas doses com possível associação a outros imunossupressores como metotrexato ou ciclofosfamida. O desenvolvimento de vasculites em pacientes com SMDs está associado a pior prognóstico da doença da base.3
Eritema elevatum diutinumO eritema elevatum diutinum (EED) é forma crônica e rara de vasculite leucocitoclástica localizada, descrita inicialmente por Hutchinson em 1888. Na descrição inicial, era dividida em duas formas: a primeira, mais comum em homens idosos; e a segunda, presente em mulheres jovens com doenças reumatológicas associadas. As duas formas atualmente são descritas na mesma entidade, e o termo diutinum significa “de longa duração” e faz referência ao caráter longo e relapsante da doença.52
As lesões clínicas apresentam‐se como pápulas, nódulos ou placas eritemato‐violáceas com predileção por superfícies extensoras dos membros.4 O acometimento cutâneo ocorre, em ordem de prevalência, no dorso das mãos, cotovelos, pernas, joelhos e pés e eventualmente região palmoplantar.52,53 Ocasionalmente, as lesões lembram vesículas, nódulos hemorrágicos ou ulcerações.53 São usualmente assintomáticas, porém podem apresentar prurido, dor ou mesmo artralgia subjacente.52 Lesões antigas com resolução parcial possuem coloração amarelada ou acastanhada, lembrando um xantoma.53
O exame histopatológico inicial demonstra vasculite leucocitoclástica que progride para fibrose perivascular com deposição de lipídeos extracelulares nos estágios avançados da doença.52 O infiltrado de polimorfonucleares ocorre em formato de cunha com deposição de fibrina na derme superficial e média.53 A ausência de leucócitos polimorfonucleares deve sugerir outro diagnóstico.53
A fisiopatologia envolve a deposição de imunocomplexos em pequenos vasos, levando a resposta inflamatória com recrutamento de leucócitos.52 As associações mais comuns são com quadros infecciosos como HIV, com frequência coinfectados com hepatites virais, CMV ou outras infecções oportunistas, além de tuberculose e infecção estreptocócica de repetição.53 Em 30% dos casos está associado a doenças autoimunes como artrite reumatoide, lúpus eritematoso sistêmico ou doença celíaca.52 Pode também estar relacionado a uma variedade de neoplasias hematológicas, particularmente a gamopatias monoclonais de isotipo IgA54 e SMDs. Mais raramente, está associado ao MM, LMC e LNH.6,55
A apresentação dermatológica pode preceder o quadro hematológico em muitos anos.55 A involução espontânea ocorre, em média 5‐10 anos após o diagnóstico.53 Entretanto, há relatos de maior duração como 25‐39 anos.55 O tratamento de escolha é feito com dapsona oral.6 Além disso, o controle da doença de base está relacionado ao controle do EED em alguns casos.55 O tratamento com medicações intralesionais ou mesmo excisão cirúrgica somente é recomendado nos casos localizados.
Pênfigo paraneoplásicoO pênfigo paraneoplásico (PPN) é variante rara de pênfigo descrita inicialmente por Anhalt et al. em 1990, que diferenciaram essa entidade do pênfigo vulgar, quando demonstraram associação com neoplasias malignas e presença de anticorpos contra desmoplaquina I e contra antígeno BP 230.56 O PPN é doença rara, com cerca de 500 casos descritos na literatura; ocorre em qualquer faixa etária, inclusive em crianças, porém a idade média é aos 64,7 anos.57,58 Alguns trabalhos aparentam tendência maior de acometimento no sexo masculino, enquanto outros mostram ausência de predileção por gênero.59–61 Em decorrência das características clínicas, histológicas e imunológicas distintas e associação a mais alta mortalidade do que os demais pênfigos, o dermatologista deve estar atento ao diagnóstico dessa patologia.
A associação com neoplasias ou desordens hematológicas é encontrada em mais de 84% dos casos. As doenças linfoproliferativas são as mais frequentemente relacionadas; o LNH é a mais comum (38,6% de todos os casos de PPN), seguida da LLC (18,4%), doença de Castleman (18,4%), timomas (5,5%), macroglobulinemia de Waldenstrom (1,2%), LH (0,6%) e gamopatia monoclonal (0,6%).57 Em geral, a neoplasia é diagnosticada antes da manifestação do PPN. Entretanto, 30% dos casos têm manifestação cutânea precedendo o diagnóstico da neoplasia oculta.62
A característica clínica proeminente do PPN é a estomatite, erosões que progridem para ulcerações orais e conjuntivais graves e refratárias que chegam a atingir toda a superfície da orofaringe com extensão para o vermelhão dos lábios.63
Dois terços dos pacientes apresentam pseudoconjuntivite membranosa grave, que pode levar a cicatrização e obliteração dos fórnices conjuntivais. Lesões na nasofaringe, esôfago, mucosas vaginal, labial, peniana e na região perianal também são encontradas.60
As lesões cutâneas geralmente surgem após as de mucosa e são polimórficas: máculas eritematosas, bolhas flácidas, erosões similares ao pênfigo vulgar (fig. 5), bolhas tensas, erupções liquenoides e lesões semelhantes ao eritema multiforme. O quadro é geralmente difuso, com predileção por tronco e extremidades proximais, acometendo mais de 50% do tegumento ou mesmo causando eritrodermia.60 A região da cabeça e face é usualmente poupada. Porém, nos pacientes que recebem radioterapia, as lesões podem se concentrar inicialmente no local irradiado com disseminação posterior, com potencial confusão com o diagnóstico com dermatite por radiação.64 Acometimento palmoplantar e ungueal com paroníquia está usualmente presente.60 A presença de lesões similares ao eritema multiforme nas palmas e plantas é útil para diferenciar do pênfigo vulgar, no qual lesões palmoplantares são incomuns.
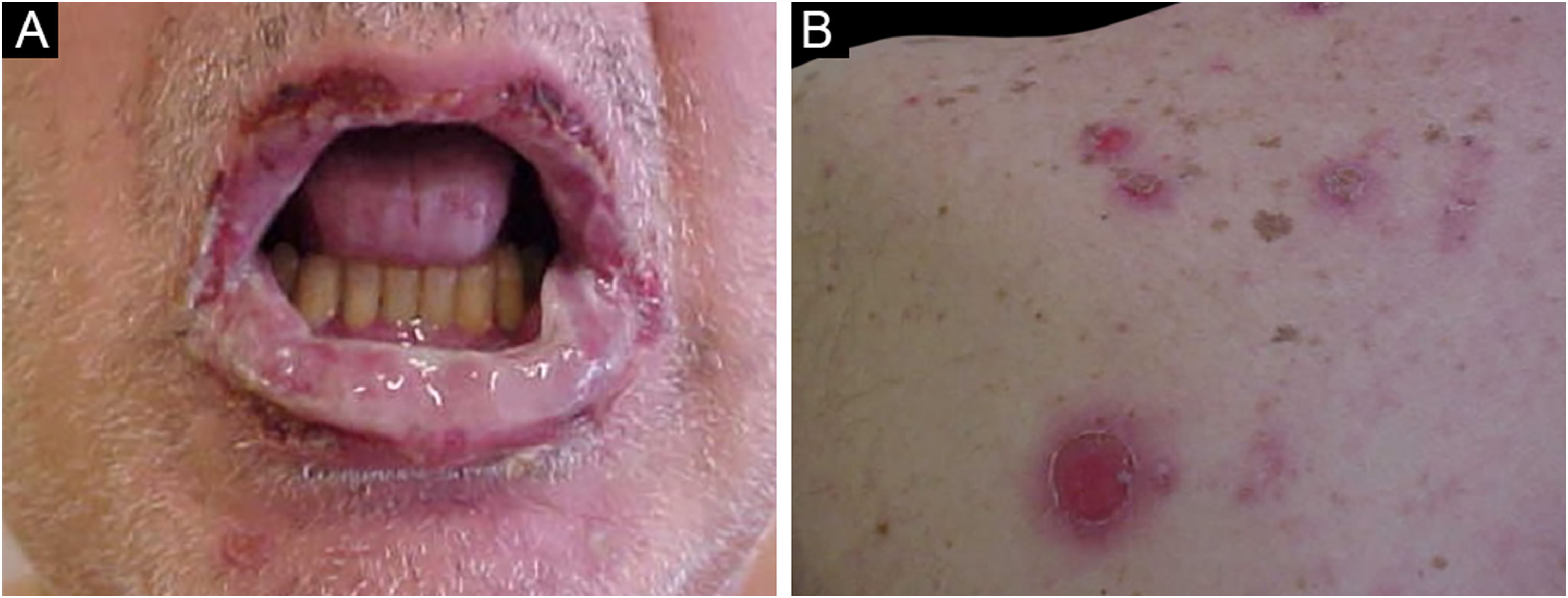
As lesões liquenoides predominam nas crianças e na população da Ásia oriental e podem ter características clínicas e histológicas semelhantes ao eritema multiforme ou doença do enxerto contra hospedeiro (DECH).59,63
Alguns pacientes desenvolvem bronquiolite obliterante, que pode levar à falência respiratória fatal, uma das principais causas de morte dos pacientes com PPN.57,65
A fisiopatologia da PPN é mais complexa que do pênfigo vulgar e ainda não está definida; respostas autoimunes humorais e mediadas por célula estão envolvidas no seu surgimento. Teorias sobre a patogênese do PPN incluem:
Surgimento de resposta imune contra antígenos neoplásicos com reação cruzada a antígenos epiteliais
Produção de anticorpos por componentes tumorais contra os antígenos epidérmicos
Desregulação do sistema imune com síntese exagerada de citocinas como IL‐6, levando a diferenciação de células B, cujos níveis estão elevados no PPN.66
Os anticorpos mais encontrados no PPN são contra envoplaquina, periplaquina e proteína α‐2‐macroglobulina‐símile 1. Raramente se encontram anticorpos IgG contra desmogleína 3 ou 1 como no pênfigo vulgar. Outros autoanticorpos encontrados são contra desmocolinas ou BP180/BP230. Para o diagnóstico sorológico, devem ser usados substratos ricos em plaquinas para a imunofluorescência indireta, como a bexiga de rato, em vez de pele humana ou esôfago de macaco.6
Em decorrência da riqueza de manifestações clínicas, a variabilidade de achados histopatológicos é alta. Em muitas lesões de estomatite são encontradas apenas inflamação, necrose e acantólise suprabasal na mucosa perilesional. As bolhas intactas apresentam acantólise suprabasal, porém, ao contrário de outros pênfigos, encontramos infiltrado inflamatório significativo mesmo nas lesões precoces. Lesões não bolhosas podem ter necrose de queratinócitos, infiltrado linfocítico na epiderme, alterações vacuolares e dermatite de interface. A imunofluorescência mostra anticorpos anti‐IgG intraepitelial, apesar de falsos negativos serem mais comuns no PPN do que no pênfigo vulgar, com necessidade de múltiplas biópsias para o diagnóstico. Uma minoria dos casos apresenta IgG positivo também na membrana basal.66
Anhalt propôs uma redefinição dos critérios diagnósticos originais em 2004, onde considera como critérios mínimos para diagnósticos de PPN com alto grau de confiança: (1) estomatite erosiva e progressiva, com envolvimento preferencial da língua; (2) achados histológicos de acantólise, alterações liquenoides ou dermatite de interface; (3) demonstração de autoanticorpos antiplaquina, que são o marcador sorológico chave do PPN; (4) demonstração de doença linfoproliferativa sobrejacente. Nos casos em que a neoplasia não está presente antes do diagnóstico, geralmente está associada a doença de Castleman, linfoma abdominal, timoma ou sarcomas retroperitoneais, detectados por tomografia de tórax, abdome e pelve.66
O diagnóstico diferencial vai depender das lesões clínicas predominantes. Em pacientes com envolvimento oral exclusivo, os diferenciais incluem pênfigo vulgar, líquen plano oral e estomatite aftosa maior. Nos pacientes com predomínio lesões liquenoides, devemos diferenciar do líquen plano e da DECH. Pacientes com lesões cutâneas e orais podem ter diagnóstico difícil entre pênfigo vulgar e PPN. Algumas características diferenciam essas duas patologias: o pênfigo vulgar apresenta acometimento oral discreto, superficial e com áreas de mucosa sã, enquanto o PPN apresenta quadro difuso, profundo e frequentemente necrótico. O pênfigo vulgar raramente afeta outras mucosas além da oral. Enquanto o pênfigo vulgar tem localização comum no couro cabeludo e poupa as áreas palmoplantares, o PPN envolve as palmas e plantas e costuma poupar o couro cabeludo. Por último, o sinal de Nikolsky é positivo no pênfigo vulgar e negativo no PPN.67
Um pequeno número de pacientes com eritema multiforme recorrente tem anticorpos positivos contra desmoplaquinas e terão imunofluorescência indireta positiva no epitélio de bexiga. Porém, diferente dos casos de PPN, esses pacientes não terão (1) malignidade subjacente; (2) anticorpos contra plaquinas mais específicas para o PPN como envoplaquina e periplaquina e (3) os anticorpos são transitórios e somente detectáveis durante os episódios da doença ativa.66
O prognóstico do PPN depende da natureza da neoplasia associada. Após o início da doença autoimune, mesmo com o tratamento e a cura da doença hematológica, existe a possibilidade de o PPN progredir. Em pacientes com neoplasias benignas ou tumores localizados, a exérese do tumor pode melhorar substancialmente ou mesmo levar o pênfigo a remissão. Em pacientes com tumores tratados por cirurgia, cerca de 50% entram em remissão. Essa remissão ocorre em um a dois anos após a cirurgia, e terapia imunossupressora normalmente é utilizada nesse período.67
Não existem ensaios clínicos randomizados sobre o tratamento do PPN em razão da raridade da doença, porém esquemas sugeridos na literatura incluem a combinação de prednisona, ciclosporina e ciclofosfamida com redução gradual das doses conforme melhora clínica.64 Outras opções terapêuticas como dapsona, azatioprina, plasmaferese, fotoferese e imunoglobulinas foram utilizados, porém nenhum mostrou eficácia relevante. As lesões cutâneas respondem mais rapidamente à terapia, enquanto a estomatite é refratária.66
Pacientes com neoplasias malignas têm respostas piores às terapias imunossupressoras, e os casos associados ao LNH parecem ter pior prognóstico que os pacientes com LLC. Quase todos os pacientes com LNH ou LLC irão a óbito entre três meses a dois anos após o diagnóstico. Corticoterapia sistêmica pode levar a melhora parcial das lesões, porém não a resolução.6
Apesar da boa resposta do rituximabe nos pacientes com pênfigo vulgar e pênfigo foliáceo, a resposta no PPN é bem menos consistente. A remissão completa é rara, e existem casos de PPN descritos em pacientes que receberam rituximabe como parte da quimioterapia da neoplasia.58
Alemtuzumabe, anticorpo monoclonal contra CD52 expresso em linfócitos T e B, mostrou‐se promissor no tratamento de alguns casos relacionados a neoplasias hematológicas, porém não impediu a progressão da doença pulmonar e está associado a imunossupressão significativa após o tratamento.58
Ictiose adquiridaA ictiose adquirida (IA), diferentemente da ictiose vulgar (IV), manifesta‐se com maior frequência na idade adulta, embora o quadro clínico seja semelhante. As manifestações clínicas incluem escamas romboides, que variam desde aspereza até descamação lamelar semelhante a escama de peixe, com bordas livres,3,49,68 coloração hipocrômica a cinza ou acastanhada, com diâmetro de menos de 1mm a mais de 1cm. Acomete principalmente o tronco e os membros, é mais acentuada nas superfícies extensoras, geralmente poupa flexuras e afeta as extremidades inferiores em maior intensidade que as extremidades superiores. As lesões cutâneas apresentam prurido, que também pode estar presente em áreas de aparência normal; escoriações secundárias ao prurido são frequentemente encontradas.49,68
Ocorre quando há interrupção no processo de cornificação, resultando em hiperceratose, descamação e anormalidades da função de barreira córnea. A patogênese é desconhecida, mas redução da síntese de lipídeos dérmicos e deficiência de colesterol são vistos frequentemente nesses pacientes e estão, provavelmente, envolvidos na patogênese.3,68
Aspectos clínicos e histopatológicos não são suficientes para diferenciar a IV da IA; a história clínica detalhada é fundamental. Essencialmente, todas as formas hereditárias de ictiose terão apresentação clínica antes dos 13 anos; em geral, há história familiar positiva e idade de início precoce, favorecendo, nesses casos, o diagnóstico de IV.49,68
Histologicamente, apresenta aumento do estrato córneo, camada granular reduzida e camada espinhosa com espessura normal. Ausência de infiltrado inflamatório na derme está entre os achados histológicos típicos. Atrofia epidérmica já foi documentada, assim como presença de leve infiltrado linfo‐histiocitário perivascular na derme papilar, sem evidência de vasculite.49,68,69
Com o diagnóstico de IA estabelecido, deve‐se avaliar o paciente quanto a possível fator desencadeante. IA pode ser resultado de doença maligna, doença não maligna ou reação medicamentosa. Se relacionada à afecção sistêmica, a manifestação cutânea ocorrer antes ou depois da identificação desta. A intensidade da IA pode estar associada à gravidade ou recidiva da causa interna.68
É frequente a associação entre IA do adulto e malignidade incluindo a doença de Hodgkin, LNH, linfoma cutâneo de células‐T (CTCL), leiomiossarcoma, micose fungoide, MM, sarcoma de Kaposi, e, raramente, a tumores sólidos, como carcinomas de ovário, mama, pulmão e colo do útero. A malignidade mais comumente relatada à IA é a doença de Hodgkin. No entanto, existem outras causas potenciais de ictiose adquirida, incluindo desnutrição, hipotireoidismo, sarcoidose e fármacos como ácido nicotínico e clofazimina.49,68
Como diagnósticos diferenciais, temos IV e doença de Refsum. A doença de Refsum deve ser suspeitada nos casos de ictiose que se apresentam da adolescência até a terceira década e estão associados a achados neurológicos como diminuição da acuidade visual, neuropatia sensorial e ataxia.68
No tratamento, hidratação e agentes ceratolíticos são úteis. A ictiose normalmente regride uma vez que a doença subjacente entre em remissão.49,68
Eritema nodosoEritema nodoso (EN) é a forma mais comum de paniculite e é caracterizado por nódulos eritematosos dolorosos, principalmente nos membros inferiores. É mais frequente em mulheres e tem pico de incidência na população com idade de 20 a 30 anos.70,71
O curso clínico do EN é caracterizado por início agudo de nódulos sensíveis e eritematosos e placas de 1–6cm de diâmetro. As lesões são bilaterais e simétricas, normalmente distribuídas nas extremidades distais inferiores nas áreas pré‐tibiais, embora as lesões também possam envolver os tornozelos, coxas e antebraços.70 Em geral, são autolimitadas, com resolução em duas a oito semanas, passando de descoloração de vermelho brilhante para descoloração amarelo‐marrom ou verde‐azulada, semelhante a hematomas. A coexistência de lesões em diferentes estágios de evolução pode ser observada no mesmo paciente. Os nódulos involuem sem ulceração, formação de cicatrizes ou atrofia, e as recorrências não são incomuns.70,71
As lesões cutâneas são frequentemente acompanhadas de sintomas sistêmicos, como febre, mal‐estar, dor de cabeça, queixas gastrintestinais (como dor abdominal, vômito e diarreia), tosse, linfadenopatia, perda de peso e artralgia, principalmente nos tornozelos e joelhos. Alguns desses achados sugerem EN secundário a doença sistêmica, e esses achados têm possibilidade de fornecer pistas diagnósticas importantes.70
A patogênese do EN é desconhecida, embora pareça ser uma resposta de hipersensibilidade a uma variedade de estímulos antigênicos. Também foi proposto que o TNF‐alfa e a IL1‐beta, liberados pelas células mielomonocíticas possam estar envolvidos.70,71
O diagnóstico geralmente é feito por meio do quadro clínico característico. No entanto, deve ser confirmado por biópsia incisional ou excisional profunda, com porção generosa de gordura subcutânea. Uma vez feito o diagnóstico patológico, o verdadeiro desafio é a identificação da etiologia subjacente, se presente, antes de considerá‐lo idiopático.4,70
Na histologia, há inflamação panicular septal predominante e ausência de vasculite primária.4
Os principais diagnósticos diferenciais são eritema induratum de Bazin, tromboflebite superficial, linfoma de células T paniculite‐símile e outras formas de paniculite. A histopatologia é essencial para diagnósticos de maior precisão.70
Embora a etiologia seja principalmente idiopática, descartar doença subjacente é fundamental antes do diagnóstico do EN. O EN pode ser o primeiro sinal de uma doença sistêmica, como infecções, doenças inflamatórias, neoplasias e medicamentos. As causas identificáveis mais comuns são infecções estreptocócicas, tuberculose primária, sarcoidose, doença de Behçet, doença inflamatória intestinal, medicamentos e gravidez. De acordo com a literatura, 32%‐72% dos casos de EN permanecem com etiologia desconhecida.4,70
Uma malignidade subjacente pode ser responsável por sintomas de EN em pacientes com quadro inexplicado e sintomas constitucionais. Malignidades hematológicas, como leucemia e linfoma, incluindo LMA, LMC e LMMC, entre outras, são mais frequentemente associadas a lesões EN, e mais raramente, neoplasias sólidas. Em alguns casos, as lesões EN indicam recidiva da doença. Pacientes com EN recorrente ou resposta fraca aos tratamentos convencionais devem ser investigados para uma malignidade subjacente.4,70
Investigação para doenças suspeitas deve ser direcionada a partir de história clínica e exame físico cuidadosos. Sintomas sistêmicos ou resultados de testes laboratoriais alterados favorecem EN secundário. A investigação laboratorial inicial deve incluir hemograma completo, velocidade de hemossedimentação (VHS), proteína C reativa, título de antiestreptolisina O, cultura de esfregaço de garganta, pesquisa de tuberculose em áreas endêmicas e radiografia de tórax. Teste de gravidez deve ser feito em todas as mulheres em idade reprodutiva.70,72
O suporte sintomático é a abordagem inicial, através de bandagens de compressão e elevação de membros com o objetivo de redução do edema e alívio da dor.70 Podem ser usados anti‐inflamatórios não esteroides para controle álgico. Corticosteroides sistêmicos, iodeto de potássio, colchicina, dapsona e hidroxicloroquina também já foram descritos.4,70
A terapia específica deve ser dedicada à condição associada subjacente, se identificada.4,72
Xantoma planoXantoma plano difuso (XPD) é doença rara, do grupo das histiocitoses não Langerhans, que acomete principalmente adultos, de ambos os sexos.73,74 A maioria dos pacientes é normolipêmica, mas ocorre também em casos de hiperlipoproteinemias dos tipos 2 a 4. A presença de xantoma cutâneo com lipídeos plasmáticos normais requer investigação adicional e monitoramento contínuo para uma condição hematológica subjacente.49,75
Apresenta‐se como máculas ou placas amarelo alaranjadas irregulares, difusas, simétricas e assintomáticas, acometendo qualquer parte do corpo, predominantemente face, tronco e áreas intertriginosas como as axilas. Geralmente, é acompanhada ou precedida por xantelasma.49,73,74
Lesões extracutâneas são possíveis de serem observadas na cavidade oral, olhos, tendões, válvula aórtica, músculos e sistema gastrintestinal.73
Os xantomas planos difusos são descritos em associação a MM, gamopatias monoclonais, e, menos frequentemente, a distúrbios linfoproliferativos.73,74
A patogênese do XPD não está claramente definida. Nos casos em que há correlação a gamopatias, alguns autores acreditam que complexos paraproteínas‐lipoproteínas sejam depositadas na pele. Outros sugerem a infiltração de células leucêmicas com subsequente xantomização.73,74
No histopatológico, estão presentes células espumosas – macrófagos com gotículas lipídicas – e número variável de células gigantes de Touton, linfócitos e histiócitos espumosos podem ser vistos na derme superior e, ocasionalmente, têm localização perivascular.73,74
Os diagnósticos diferenciais incluem xantomas hiperlipidêmicos e xantogranuloma necrobiótico.73,74
O prognóstico depende da condição subjacente, e o tratamento deve ser direcionado para a doença de base. A remissão da condição hematológica resulta em melhora cutânea, com recidiva indicada por recorrência de lesões na pele.49,73
Xantogranuloma necrobióticoO xantogranuloma necrobiótico (XGN) é histiocitose incomum de células não Langerhans, que envolve a pele e tecidos extracutâneos, com predileção pela região periorbitária.76,77 Pode envolver o olho, ocasionando proptose, blefaroptose e limitação da motilidade ocular.77 Órgãos internos também podem ser acometidos, incluindo os tratos gastrintestinal e respiratório.77–79 Raros casos apresentam lesões no tronco ou extremidades, sem envolvimento facial.80
As lesões do XGN iniciam‐se mais frequentemente pela região periorbitária, progredindo para o tronco e as extremidades.77,78
É doença caracterizada por múltiplas pápulas e nódulos endurecidos, assintomáticos, amarelados a marrom‐avermelhados, que evoluem lentamente para grandes placas envolvendo a derme e o tecido subcutâneo.77 Ulceração, telangiectasias, atrofia e prurido também podem estar presentes e são mais observados na região periorbitária.77,78
A idade média de início é a sexta década de vida, com casos descritos na faixa etária de 17 a 85 anos, não havendo predileção por sexo.80
Em aproximadamente 80% dos pacientes, é identificada gamopatia monoclonal, resultante de discrasia de células plasmáticas ou de um distúrbio linfoproliferativo.76–78 Foi observado que apenas 10% dos casos evoluem para MM.80
Outras associações relatadas incluem LNH, LH, macroglobulinemia de Waldenstrom, SMD, LLC e amiloidose.77,80,81 Todos os pacientes devem ser investigados para distúrbios hematológicos e linfoproliferativos.77
O mieloma múltiplo que se desenvolve em pacientes com XGN parece apresentar um comportamento relativamente benigno, com 90%‐100% dos pacientes apresentando sobrevida de 10‐15 anos.80
A patogênese da XGN ainda não foi totalmente esclarecida.77
XGN e xantoma plano (XP) são duas formas distintas de xantoma; casos que relataram a coexistência de XGN e XP ao mesmo tempo foram raros.82 Se uma nova lesão XGN se desenvolver em um paciente com XP, uma avaliação da doença hematológica é fortemente recomendada.82
No exame histopatológico podem ser observados epiderme atrófica ou normal, extensas áreas de necrobiose e infiltrado granulomatoso com células gigantes de corpo estranho, células gigantes de Touton e células espumosas.76,77 Embora nenhum padrão histológico seja específico para XGN, essas alterações, associadas ao quadro clínico característico, favorecem o diagnóstico de XGN.76,77
As opções de tratamento incluem corticosteroides tópicos e sistêmicos, talidomida, imunoglobulina intravenosa de alta dose (IVIG), clorambucil, ciclofosfamida, fludarabina, rituximabe, melfalano, infliximabe, interferon alfa, cladribina, hidroxicloroquina, azatioprina, metotrexato, terapia a laser, radioterapia, cirurgia, PUVA, plasmaférese e fotoférese extracorpórea, com respostas variáveis e imprevisíveis.76,77,79
O tratamento da gamopatia monoclonal com agentes alquilantes não influencia necessariamente a atividade da doença de pele.77,78
O XGN tem um curso crônico, progressivo e indolente. O prognóstico depende da gravidade das manifestações extracutâneas, da presença de neoplasias hematológicas e das complicações das lesões cutâneas.80
EscleromixedemaEscleromixedema é doença rara que atinge, principalmente, adultos entre 30 e 80 anos, sem predomínio de raça ou sexo. Raramente relatada em bebês e crianças pequenas.83,84 A condição geralmente está associada a gamopatias monoclonais. A doença progride para MM definitivo em menos de 10% dos casos. Associação com outras doenças hematológicas malignas, como LH e LNH, macroglobulinemia Waldenström e LMMC ou carcinomas viscerais já foram relatados.49,83
Manifesta‐se como uma erupção generalizada de pápulas de 2–3mm, de aspecto firme e céreo, pouco espaçadas, em forma de cúpula ou planas, envolvendo mãos, antebraços, cabeça, pescoço, tronco e coxas superiores.49,83 As pápulas são, frequentemente, organizadas em disposição linear e a pele ao redor é brilhante e endurecida (esclerodermoide). Raramente, nódulos subcutâneos estão presentes. A glabela é tipicamente envolvida, com sulcos longitudinais profundos que produzem face de aspecto leonino característico.83 Sulcos profundos ocasionalmente são evidenciados no tronco ou membros, produzindo o “sinal de Shar‐Pei”. Eritema, edema e coloração acastanhada ocorrem nas áreas não envolvidas; o prurido é comum. Sobrancelhas, pelos axilares e púbicos eventualmente são esparsos nesses pacientes. As membranas mucosas são poupadas. Enquanto a doença progride, surgem placas eritematosas e infiltradas com consequente enrijecimento da pele, esclerodactilia e diminuição da motilidade da boca e articulações. Sobre a articulação interfalangiana proximal, uma placa com depressão central e borda elevada (devido ao espessamento da pele) pode ser vista e é referido como o “sinal de donut”.49,83
Em pacientes com escleromixedema, as manifestações extracutâneas mais comuns são anormalidades neurológicas, reumatológicas e anormalidades cardíacas.83
A patogênese do escleromixedema é desconhecida. A principal hipótese é de que citocinas circulantes, como IL‐1, TNF‐alfa e TGF‐beta, estimuladoras da síntese de glicosaminoglicanos e proliferação de fibroblastos na pele, possam desempenhar um papel no desenvolvimento das lesões. A remissão clínica do escleromixedema após o transplante autólogo de células‐tronco hematopoéticas (TCTH) sugere que a medula óssea possa ser fonte dessas citocinas. Apesar da associação frequente a gamopatia monoclonal, os níveis de paraproteína geralmente não se correlacionam com a gravidade da doença, a progressão da doença ou a resposta ao tratamento.83
O diagnóstico de escleromixedema é feito com base nos seguintes critérios clínico‐patológicos:
- •
Erupção papular e esclerodermoide generalizada/difusa;
- •
Tríade de deposição de mucina (composto principalmente de ácido hialurônico na parte superior e derme reticular média), proliferação de fibroblastos e fibrose;
- •
Gamopatia monoclonal;
- •
Ausência de distúrbio da tireoide.83,85,86
Os principais transtornos a serem considerados no diagnóstico diferencial são escleredema, esclerodermia localizada, esclerose sistêmica, fibrose sistêmica nefrogênica e líquen mixedematoso.83,85
As opções terapêuticas incluem intervenções direcionadas ao tratamento da discrasia de células plasmáticas associada.85,86
Em decorrência da raridade da doença, não há tratamento específico eficaz. A terapia de primeira linha é feita com imunoglobulina intravenosa (IVIg), na dose de 2 g/kg, dividida em dois a cinco dias consecutivos, aplicada mensalmente.87,88 O mecanismo pelo qual IVIg age no escleromixedema é pouco claro, mas deve ser considerado particularmente em pacientes com deterioração dos sintomas cutâneos ou envolvimento de órgãos internos com risco de vida.83–85
Quando o tratamento com IVIg não é indicado ou quando ocorre resposta insuficiente, talidomida (ou lenalidomida) e glicocorticoides sistêmicos são as opções da próxima linha de tratamento. Talidomida e glicocorticoides sistêmicos são administrados sozinhos ou em combinação com IVIg.81,85
Outras estratégias direcionadas à pele, como fotoferese extracorpórea, PUVA, feixe de elétrons, corticosteroides tópicos e retinóides foram tentados com sucesso variado.83,85 Pacientes podem se beneficiar de intervenções direcionadas ao tratamento da discrasia de células plasmáticas associada.85
ConclusãoComo revisado em ambos os artigos (parte I e parte II), as manifestações cutâneas antecedem, acompanham ou são tardias ao diagnóstico das neoplasias hematológicas. Desse modo, o reconhecimento dessas entidades e sua possível relação com uma provável doença de base tem um significante valor diagnóstico.
A importância do reconhecimento das doenças dermatológicas relacionadas às neoplasias hematológicas se faz presente, pois o acometimento cutâneo piora a morbidade do paciente que necessita de tratamento dermatológico específico. Além disso, a presença da doença de pele pode configurar piora no prognóstico da afecção hematológica de base e possivelmente desencadeia a necessidade de mudança no aporte terapêutico sistêmico do doente.
Tudo que foi discutido nesta revisão evidencia a relevância de se conhecer as manifestações cutâneas que acompanham quadros sistêmicos, seja pelo simples cuidado a ser oferecido ao doente e consequente manutenção da qualidade de vida, mas principalmente por alertar precocemente a existência de uma neoplasia oculta ou a piora de uma já existente.
Nos doentes oncológicos, as comorbidade se somam, e o diagnóstico diferencial dermatológico é um desafio. Devem‐se considerar etiologias neoplásicas, inflamatórias, infecciosas, além das farmacodermias. As manifestações cutâneas das doenças hematológicas são extremamente variáveis na morfologia, distribuição e sintomatologia. A biópsia de pele e exame imuno‐histoquímico colaboram substancialmente ao diagnóstico clínico, assim como os dados microbiológicos dos fragmentos teciduais enviados a culturas para fungos e bactérias.
Poucos estudos evidenciam as doenças de pele que acompanham o doente hematológico e pouco se conhece sobre os mecanismos que levam a esses quadros, sendo um amplo e importante campo de estudo. Também é pouco conhecido quanto essas enfermidades impactam na qualidade de vida dos pacientes hematológicos e quanto elas funcionam como marcadores prognósticos.
O manejo dessas entidades não é simples e retrata a importância de cada passo ser discutido de modo multidisciplinar. A condução da intervenção dermatológica conjunta ao hematologista sempre se impõe. Entre muitos fatos que devem ser observados está quanto o tratamento da doença de base será resolutivo ao quadro dermatológico e quanto a terapêutica dermatológica específica impacta positiva ou negativamente o contexto geral.
Cabe ao dermatologista estar presente junto à equipe hematológica no sentido de esclarecer o possível diagnóstico do quadro cutâneo, planejar conjuntamente o tratamento, manter o seguimento e, acima de tudo, fornecer o melhor cuidado ao paciente. Cabe ao hematologista observar a pele do paciente e solicitar a intervenção dermatológica para melhor condução diagnóstica. Na medicina atual, é primordial a importância do cuidado integral, multidisciplinar/colaborativo, excelente tecnicamente e amplamente humanizado.
Suporte financeiroNenhum.
Contribuição dos autoresPatricia Karla de Souza: Concepção do estudo; redação do artigo e revisão crítica do conteúdo intelectual importante; desenho do estudo em conjunto com coautores; revisão crítica da literatura; aprovação final da versão final do manuscrito.
Rafael Oliveira Amorim: Redação do artigo e revisão crítica do conteúdo intelectual importante; desenho do estudo em conjunto com coautores; revisão crítica da literatura; aprovação final da versão final do manuscrito.
Letícia Siqueira Sousa: Redação do artigo e revisão crítica do conteúdo intelectual importante; desenho do estudo em conjunto com coautores; revisão crítica da literatura; aprovação final da versão final do manuscrito.
Mariana Dias Batista: Redação do artigo e revisão crítica do conteúdo intelectual importante; desenho do estudo em conjunto com coautores; revisão crítica da literatura; aprovação final da versão final do manuscrito.
Conflito de interessesNenhum.
Como citar este artigo: Souza PK, Amorim RO, Sousa LS, Batista MD. Dermatological manifestations of hematologic neoplasms. Part II: nonspecific skin lesions/paraneoplastic diseases. An Bras Dermatol. 2023;98:141–58.
Trabalho realizado no Hospital Israelita Albert Einstein, São Paulo, SP, Brasil e no Departamento de Dermatologia, Universidade Federal de São Paulo, São Paulo, SP, Brasil.
