







A associação entre penfigoide bolhoso (PB) e lesões de milia tem sido cada vez mais relatada, mas sua prevalência ainda não havia sido relatada em uma população brasileira com PB.
ObjetivosDescrever a ocorrência e os achados clínico‐laboratoriais da associação PB‐milia em uma amostra da região sudeste brasileira.
MétodosRealizou-se estudo descritivo baseado nos prontuários de 102 pacientes com PB. Dados clínicos e laboratoriais de pacientes com PB‐milia foram obtidos. IgE sérica total, ensaios de immunoblotting com base em antígenos da zona da membrana basal e tipagem de alelos HLA‐DQ foram realizados.
ResultadosLesões de milia foram evidentes em oito (7,8%) pacientes com PB, cinco do sexo masculino, com idade entre 46 e 88 anos. Níveis elevados de IgE total foram determinados em sete (87,5%) de oito pacientes. Em cinco dos oito pacientes, o immunoblotting mostrou reatividade de IgG contra o domínio BP180‐NC16a, mas não contra colágeno VII ou laminina‐332; também revelou reatividade contra o domínio C‐terminal de BP180 ou LAD‐1, ou ambos em quatro deles. Os alelos HLA‐DQB1*03:01 e HLA‐DQA1*05:05 foram identificados em três desses cinco pacientes com PB‐milia. Além disso, três dos cinco casos apresentaram o grupo alélico HLA-DQB1*06.
Limitações do estudoO HLA foi determinado em cinco pacientes.
ConclusõesA formação de milia em pacientes com PB parece ser menos incomum do que anteriormente admitido. Os dados laboratoriais revelaram aumento de IgE; autoanticorpos contra o domínio C‐terminal de BP180 ou LAD‐1, ou ambos; e o grupo alélico HLA-DQB1*06, descrito na associação PB‐milia. A determinação cuidadosa de anticorpos contra moléculas da zona da membrana basal e a caracterização do HLA em diferentes populações podem fornecer mais informações sobre essa associação.
O penfigoide bolhoso (PB) é a doença bolhosa autoimune mais prevalente no mundo. Caracteriza‐se clinicamente por prurido, bolhas tensas e lesões urticariformes na pele; vesículas e erosões nas membranas mucosas são menos frequentes. Bolhas subepidérmicas com infiltrado inflamatório eosinofílico predominante na derme superior são evidentes na histopatologia. Autoanticorpos contra as proteínas hemidesmossômicas BP180 (também denominadas BPAG2 ou colágeno tipo XVII) ou BP230 (também denominadas BPAG1 ou distonina), ou ambos, são demonstrados em amostras de soro.1
O PB é conhecido por estar associado a alelos HLA e fatores ambientais.2–4 É interessante notar que sua apresentação clínica tem sido associada a milia nas cicatrizes de lesões bolhosas ou na pele não lesionada em certos pacientes com PB.5 A presença de milia está associada principalmente à epidermólise bolhosa adquirida (EBA) e porfiria cutânea tardia.6,7 Prost et al. (1987) descreveram a presença de milia em casos de EBA e penfigoide cicatricial, mas lesões de milia estavam ausentes nos pacientes analisados com PB.8 Relatos sobre a associação PB‐milia vêm aumentando nas últimas décadas em populações diversas, mas sua prevalência e dados clínico‐laboratoriais ainda não foram relatados na população brasileira com PB.9–15
Dos 102 casos de PB compilados, relatamos oito pacientes que apresentaram associação com milia. Avaliamos os achados clínicos, laboratoriais e de immunoblotting e determinamos os alelos HLA‐DQ nesses oito pacientes com PB‐milia.
MétodosEste estudo descritivo foi aprovado pelo Comitê de Ética local (n° 12248/2010).
Foram revisados os prontuários de 102 pacientes diagnosticados com PB em um período de 35 anos. Todos eles moravam na região sudeste do Brasil. O diagnóstico de PB foi baseado em achados clínicos, confirmados pela presença de bolhas subepidérmicas ao exame histopatológico e por fluorescência linear de IgG na zona da membrana basal (ZMB) por imunofluorescência direta e indireta (IFD e IFI, respectivamente). A IFI com anti‐IgA e anti‐IgG foi realizada pelo método salt‐split skin (SSS). Os autoanticorpos IgG anti‐BP180 e anti‐BP230 foram determinados por ensaios ELISA (valor de corte=9 U/mL; MBL, Japão).
Os dados clínicos foram avaliados para os oito pacientes com PB‐milia incluídos neste estudo, e ensaios laboratoriais foram realizadas em amostras de DNA e soro armazenados. Para a determinação de IgE total, 50μL de soro foram preparados de acordo com as recomendações do fabricante (ImmunoCAPTM Total IgE, Thermo‐Fisher, EUA). A titulação foi realizada em Phadia100 (Thermo‐Fisher, EUA). Foram realizados os testes de immunoblotting (IB) com antígenos ZMB: e com os domínios C‐terminais BP180‐NC16a e BP180, moléculas de BP230, LAD‐1, colágeno tipo VII e laminina‐332. Os alelos HLA‐DQ foram determinados por PCR combinada com sondas oligonucleotídicas sequência‐específicas (PCR‐SSOP). O kit LAB Type (One Lambda Inc., Kittredge, CA, EUA) foi utilizado de acordo com as instruções do fabricante e a tecnologia Luminex.
ResultadosDos 102 casos de PB compilados, oito apresentaram lesões de milia (figs. 1 e 2), correspondendo à prevalência de PB‐milia de 7,8%. A tabela 1 resume os dados demográficos e clínicos desses oito pacientes. Cinco eram do sexo masculino; a mediana da idade era de 75,5 anos; e a mediana da duração da doença era de cinco meses. A descrição clínica mostrou lesões ativas de PB nos oito pacientes. Apenas um paciente apresentou acometimento mucoso. Nenhum deles havia mudado sua medicação habitual antes do início do PB. Digno de nota, quatro pacientes apresentavam doenças neurológicas (DN). Sete pacientes responderam bem ao tratamento usual, e um faleceu em decorrência de tromboembolismo pulmonar após cirurgia de hérnia de disco.
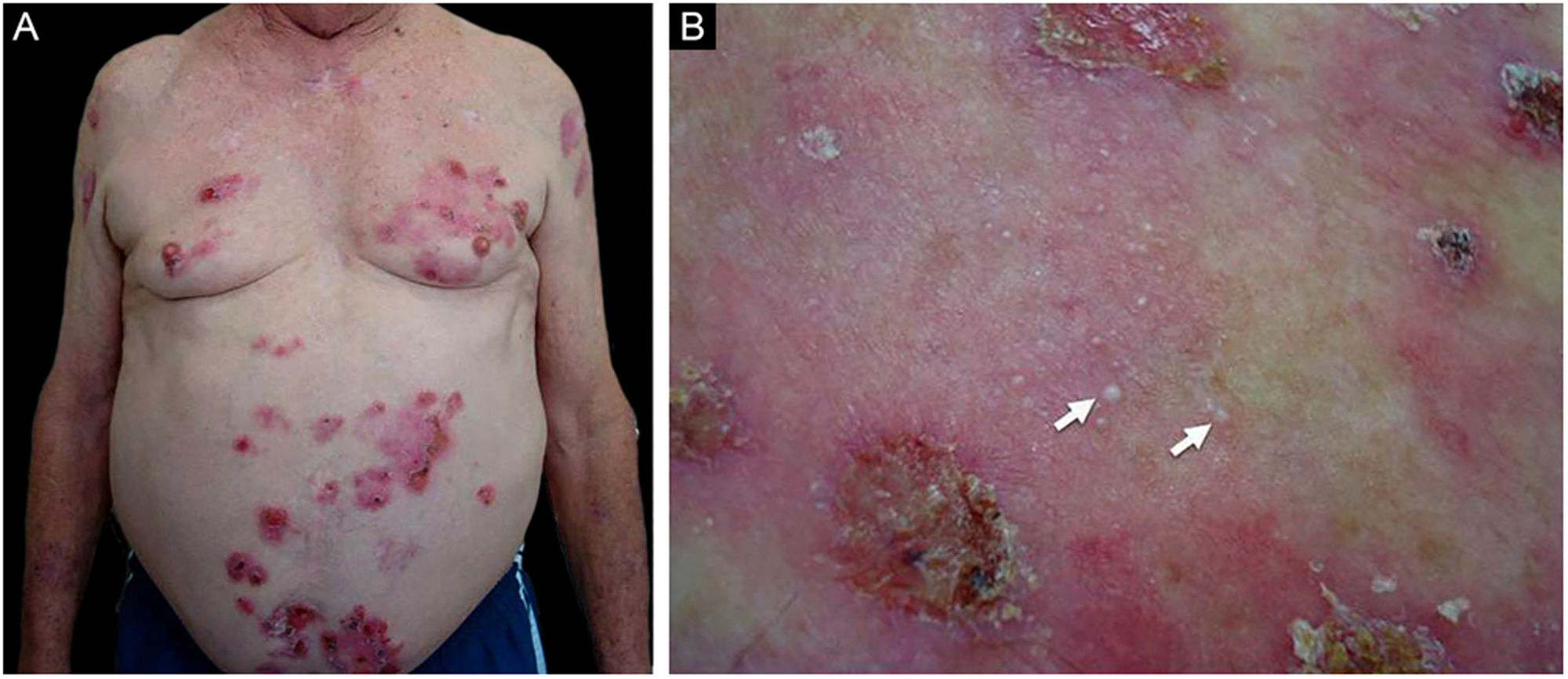
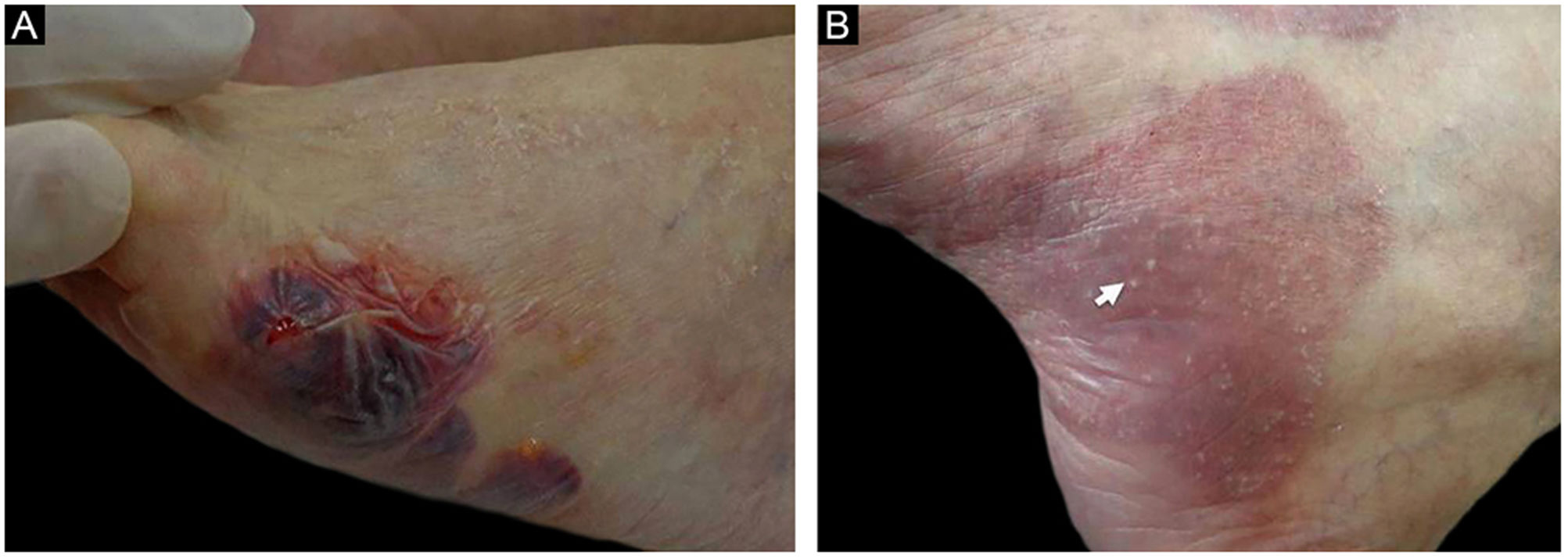
Dados demográficos e clínicos de pacientes com associação PB‐milia
| Número do paciente | Sexo | Idade (anos) | Duração da doença (meses) | Distribuição das lesões | Histórico médico e comorbidades | Medicamentos em uso no início das lesões de PB |
|---|---|---|---|---|---|---|
| 1 | Masculino | 83 | 5 | Tronco, membros, mãos e pés | Erisipela recorrente, hipertensão, dislipidemia e câncer de bexiga ressecado | Enalapril, amlodipina, sinvastatina, carvedilol e omeprazol |
| 2 | Masculino | 71 | 10 | Tronco, abdome e membros | Doença da tireoide, hipertensão, diabetes mellitus tipo 2 e história de histoplasmose pulmonar | Metformina, losartana, aspirina, ramipril, omeprazol e atorvastatina |
| 3 | Masculino | 85 | 6 | Tronco e membros inferiores | Hipertensão, diabetes mellitus tipo 2 e acidente vascular encefálico | Aspirina, carvedilol, furosemida, dinitrato de isossorbida, omeprazol, NPH e insulina |
| 4 | Feminino | 67 | 3 | Pescoço, tronco e membros | Hipertensão, hipotireoidismo, hiperparatireoidismo e asma | Losartana, amlodipina, levotiroxina, ranitidina, duloxetina, pregabalina, primidona, carbonato de cálcio, pidolato de magnésio, cloreto de potássio, ácido fólico e vitamina E |
| 5 | Masculino | 80 | 4 | Tronco e mãos | Hipertensão, diabetes mellitus tipo 2, dislipidemia e acidente vascular encefálico | Hidroclorotiazida, atenolol, aspirina, fenitoína, metformina, sinvastatina, diosmina, indapamida e cilostazol |
| 6 | Masculino | 70 | 8 | Couro cabeludo, face, tronco, abdômen e membros superiores | Hipertensão, dislipidemia, tabagismo, alcoolismo, infecção urinária de repetição, acidente vascular encefálico e episódio de psicose | Hidroclorotiazida, sinvastatina, clopidogrel, nortriptilina e amiodarona |
| 7 | Feminino | 46 | 3 | Região axilar, coxas e pés | Cefaleia crônica | Desogestrel, etinilestradiol e metamizol |
| 8 | Feminino | 88 | 5 | Mucosa oral, tórax, mãos e pés e região inguinal | Doença de Parkinson e demência | Prolopa, domperidona, omeprazol, doxiciclina e nicotinamida |
A tabela 2 detalha os achados laboratoriais. Seis dos sete pacientes apresentavam bolha subepidérmica, e infiltrado inflamatório eosinofílico na derme superior predominou em quatro pacientes ao exame histopatológico. Em cinco desses pacientes, a IFD confirmou IgG ou C3, ou ambos, mas nenhuma fluorescência linear de IgA na ZMB (fig. 3A). Em oito pacientes, SSS‐IFI demonstrou IgG ou C3, ou ambos, mas nenhuma fluorescência linear de IgA no lado epidérmico da clivagem (fig. 3B). De interesse, o aumento dos níveis séricos de IgE total variando de 252 a >5.000 kU/mL foi documentado em sete dos oito pacientes. Entre os oito pacientes, o teste ELISA mostrou níveis aumentados de anticorpos anti‐BP180, anti‐BP230 e em ambos anti‐BP180 e anti‐BP230 em dois, um e três pacientes, respectivamente; dois pacientes testaram negativo para os anticorpos anti‐BP180 e anti‐BP230.
Dados laboratoriais de pacientes com associação PB‐milia
| Número do paciente | Características histopatológicas na biópsia de pele | IFD | SSS‐ IFI | IgE sérico (kU/mL)a | Anti‐BP180 (U/mL)b | Anti‐BP230 (U/mL)b | Achados de Immunoblotting | AlelosHLA‐DQ |
|---|---|---|---|---|---|---|---|---|
| 1 | Bolha subepidérmica com eosinófilos dentro da bolha e na derme superior | IgG e C3 | IgG e C3 | > 5000 | 128,9 | 137,9 | Domínio BP180‐NC16a | N.D. |
| 2 | Descolamento epidérmico; infiltrado inflamatório com linfócitos, neutrófilos e eosinófilos na derme superior | C3 e fibrinogênio; IgG negativo | IgG e C3 | > 5000 | 2,3 | 4,6 | Domínio C‐terminal BP180 e LAD‐1 | N.D. |
| 3 | Epiderme exibindo leve espongiose. Discreto infiltrado inflamatório linfocitário intersticial na derme superior | Negativo | IgG | 782 | 5,3 | 1,7 | Nenhum | N.A. |
| 4 | Foco de descolamento epidérmico. Infiltrado inflamatório perivascular e intersticial leve com linfócitos na derme superior | IgG e C3 | IgG e C3 | > 5000 | 159,2 | 72,0 | Domínio BP180‐NC16a | DQA1* 01:02/02:01 |
| DQB1* 02:02/06:02 | ||||||||
| 5 | N.D. | IgG e C3 | IgG | 252 | 107,3 | 4,5 | BP180‐NC16a, domínio C‐terminal BP180, e LAD‐1 | DQA1* 01:01/05:05 |
| DQB1* 03:01/05:01 | ||||||||
| 6 | Vesícula subepidérmica com fibrina, neutrófilos e linfócitos. Moderado infiltrado linfocitário perianexial, perivascular e intersticial na derme superior | Negativo | IgG | 1286 | 4,4 | 68,1 | BP230; domínio C‐terminal BP180 | DQA1* 01:02/02:01 |
| DQB1* 02:02/06:02 | ||||||||
| 7 | Vesícula subepidérmica; infiltrado inflamatório com numerosos eosinófilos e linfócitos na derme superior | N.D. | IgG | 7,1 | 98,4 | 1,0 | BP180‐NC16a, domínio C‐terminal BP180, e LAD‐1 | DQA1* 05:05/05:05 |
| DQB1* 03:01/03:01 | ||||||||
| 8 | Vesícula subepidérmica com alguns eosinófilos em seu interior; ceratinócitos necróticos no assoalho da clivagem. Infiltrado inflamatório intersticial e perivascular com predomínio de linfócitos e eosinófilos na derme superior | Negativo | IgG e C3 | 427 | 169,2 | 23,3 | Domínio BP180‐NC16a e LAD‐1 | DQA1* 01:03/05:05 |
| DQB1* 03:01/06:03 |
PB, penfigoide bolhoso; IFD, imunofluorescência direta com deposição de fluorescência linear ao longo da zona da membrana basal; SSS‐IFI, método salt‐split skin na imunofluorescência indireta com fluorescência linear na clivagem epidérmica; fluorescência IgA negativa. N.D., não disponível.
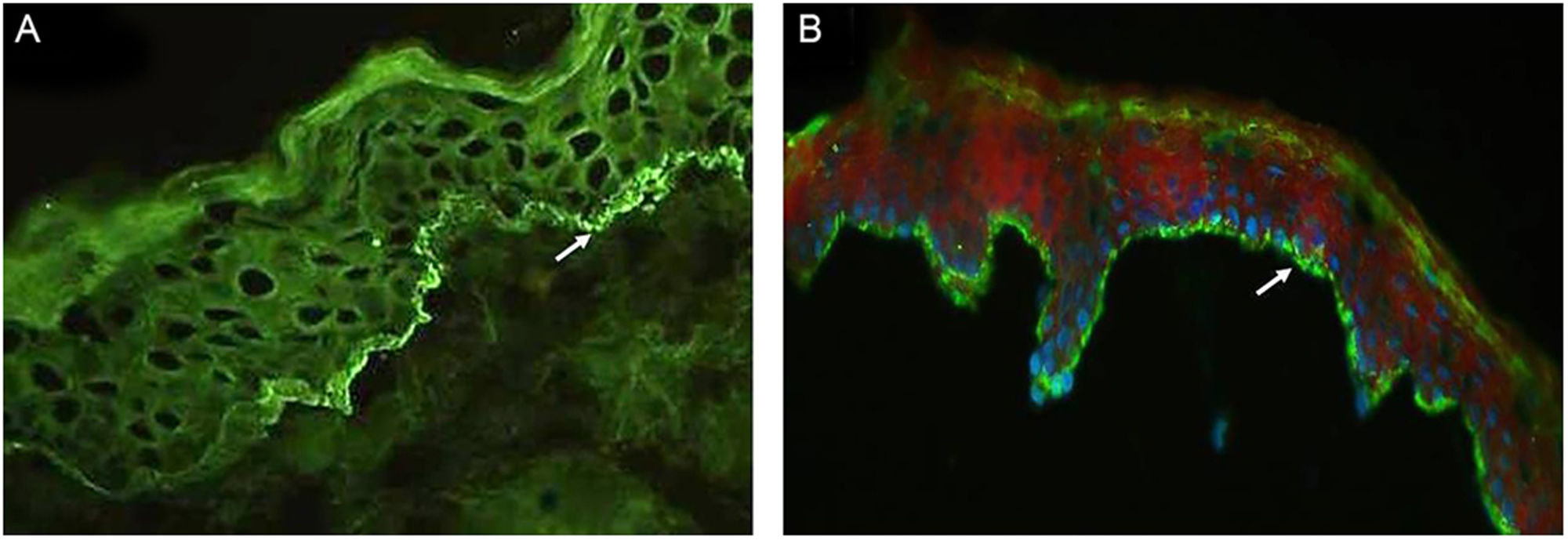
(A), Imunofluorescência direta em biópsia de pele mostrando fluorescência linear de IgG na zona da membrana basal (seta branca; paciente 2). Imunofluorescência, 400×. (B), Imunofluorescência indireta utilizando 1M‐NaCl‐salt‐split skin mostra fluorescência de IgG no lado epidérmico de clivagem (paciente 5). Imunofluorescência, 400×.
Entre os oito pacientes, os testes de IB destacaram a reatividade de IgG contra BP230 em um paciente (dados não mostrados), contra o domínio BP180‐NC16a em cinco pacientes (fig. 4A) e contra o domínio C‐terminal de BP180 ou LAD‐1, ou ambos, em quatro pacientes (figs. 4B e C). Nenhum paciente apresentou reatividade contra colágeno tipo VII ou laminina‐332. Os alelos HLA‐DQ foram definidos em cinco dos oito pacientes (ver tabela 2, última coluna).
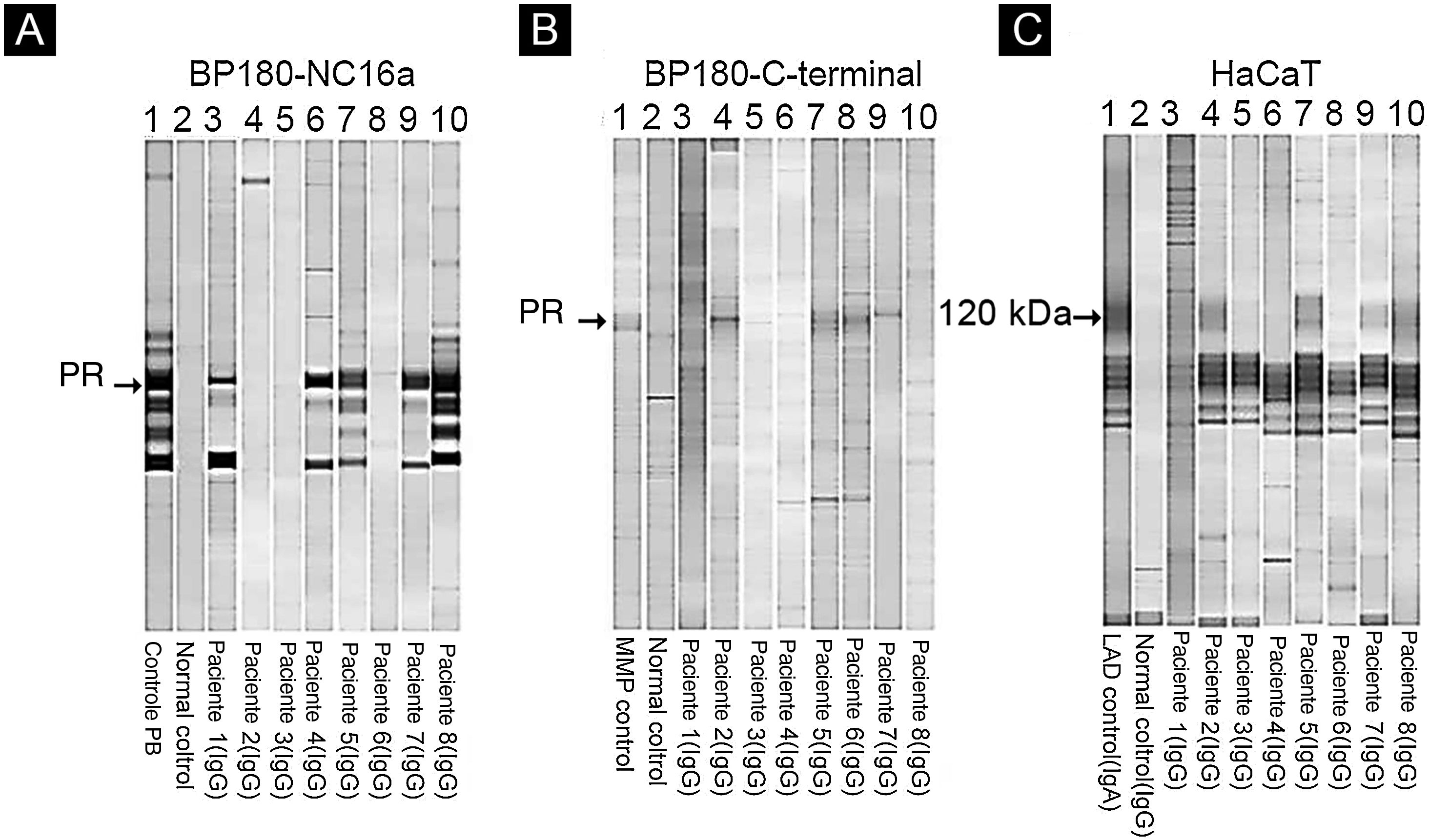
Immunoblotting utilizando BP180‐NC16a (A) C‐terminal BP180 (B) e células HaCaT (C) com amostras de soro. Para resultados positivos, procure a proteína de referência (PR) de: soro do paciente PB (penfigoide bolhoso) (A), MMP, soro do paciente com penfigoide das membranas mucosas (B) e LAD: soro do paciente com dermatose IgA linear (C).
Que seja de nosso conhecimento, a associação PB‐milia ainda é considerada um achado clínico incomum.5,9–15 Não há relato da associação PB‐milia na população brasileira. Aqui, é apresentada a prevalência de 7,8% de associação PB‐milia em 102 pacientes com PB, indicando que o diagnóstico diferencial cuidadoso com EBA é obrigatório, visto que a formação de milia é frequentemente relatada em EBA.6
Em virtude do aumento do dos relatos da associação PB‐milia, é interessante destacar algumas características clínicas e laboratoriais apresentadas pelos oito pacientes PB‐milia: (i) Associação com DN foi descrita em quatro pacientes ‐ três com acidente vascular cerebral (pacientes 3, 5 e 6) e um com doença de Parkinson e demência (paciente 8). DN em associação com PB estabelece uma relação interessante para a patogênese do PB,4,16 mas nenhuma associação DN‐PB‐milia foi encontrada na literatura. (ii) Níveis de IgE total aumentados foram demonstrados em sete pacientes. O aumento da IgE total foi relacionado à gravidade do PB,17–20 e foi relatado em um caso de associação de PB‐milia.13 O fato do aumento da IgE total ter sido identificado em 87,5% dos pacientes com PB‐milia aqui incluídos merece confirmação futura. (iii) Curiosamente, quatro pacientes apresentaram anticorpos contra o domínio C‐terminal de BP180 e LAD‐1 em IB. A interação de proteínas hemidesmossômicas e componentes da matriz extracelular sob os hemidesmossomos também pode resultar na formação de milia,5 mas essas características não são exclusivas da associação PB‐milia.
Por fim, os genes HLA são provavelmente o fator de predisposição genética mais significativo na patogênese da PB.2 Na maior série de casos de associação PB‐milia até o momento, lesões de milia estavam presentes em 23 (31,1%) dos 74 pacientes britânicos com PB, e o grupo alélico HLA‐DQ6 foi associado à condição PB‐milia.21 Aqui, três dos cinco casos apresentavam o grupo alélico HLA‐DQB1*06. Além disso, três exibiam os alelos HLA‐DQA1*01:03/05:05, o alelo HLA‐DQB1*03:01, ou todos eles, que representam alelos de suscetibilidade ao PB na população brasileira.22
ConclusãoEm conclusão, lesões de milia parecem ser uma descrição clínica atual associada ao PB, configurando um diagnóstico diferencial especial com outras doenças bolhosas. A formação de milia em PB merece uma explicação baseada na patogênese do motivo pelo qual certos pacientes com PB as desenvolvem.
Suporte financeiroEste estudo foi parcialmente financiado pela FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo), processo número 2010/51729‐2, e pela FAEPA (Fundação de Apoio ao Ensino, Pesquisa e Assistência). SV e TAJ receberam bolsa de doutorado da CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) e da FAPESP, respectivamente.
Contribuição dos autoresSebastián Vernal: Contribuiu com a obtenção e interpretação dos dados, redação do manuscrito, participação efetiva, revisão da literatura, aprovação da versão final do manuscrito.
Ederson Valei de Oliveira: Contribuiu com a obtenção e interpretação dos dados, participação efetiva, aprovação da versão final do manuscrito.
Roberto Bueno Filho: Contribuiu com a obtenção e interpretação dos dados, participação efetiva, participação em conduta terapêutica dos casos estudados, aprovação da versão final do manuscrito.
Tamiris A. Julio: Contribuiu com a obtenção e interpretação dos dados, participação efetiva, aprovação da versão final do manuscrito.
Eduardo A. Donadi: Contribuiu com a obtenção e interpretação dos dados, participação efetiva, aprovação da versão final do manuscrito.
Aline Turatti: Contribuiu com a obtenção e interpretação dos dados, participação efetiva, aprovação da versão final do manuscrito.
Norito Ishii: Contribuiu com a obtenção e interpretação dos dados, participação efetiva, aprovação da versão final do manuscrito.
Takashi Hashimoto: Contribuiu com a obtenção e interpretação dos dados, participação efetiva, redação do manuscrito, aprovação da versão final do manuscrito.
Ana Maria Roselino: Contribuiu com a concepção e planejamento do estudo, obtenção e interpretação dos dados, redação do manuscrito, participação efetiva, revisão da literatura, participação em conduta terapêutica dos casos estudados, aprovação da versão final do manuscrito.
Conflito de interessesNenhum.
Os autores agradecem a Neifi Deghaide pela assistência técnica na determinação do HLA, à professora Karla Arruda por permitir o uso do laboratório para a dosagem de IgE total e ao corpo clínico do ambulatório de dermatoses autoimunes da Divisão de Dermatologia do Hospital Universit´rio da Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Brasil.
Como citar este artigo: Vernal S, Oliveira EV, Bueno Filho R, Julio TA, Donadi EA, Turatti A, et al. Bullous pemphigoid and milia: prevalence and clinical laboratory findings in a Brazilian sample. An Bras Dermatol. 2022;97:435–42.
Trabalho realizado no Hospital Universitário, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brasil e na Kurume University Institute of Cutaneous Cell Biology, ßukuoka, Japão.
