




Dear Editor,
In 1893, Mibelli and Respighi coined the term porokeratosis assuming that the columns of parakeratosis emerged from the ostia of eccrine ducts. Later on, it turned out to be a misnomer when lesions were proved to result from a clonal disorder of keratinization characterized by the appearance of atrophic patches surrounded by the cornoid lamella.1,2 Although the disorder is inherited in an autosomal dominant trait with incomplete penetrance, most cases are caused by sporadic mutations.3,4
Porokeratosis subtypes include the classic porokeratosis of Mibelli, disseminated superficial porokeratosis, disseminated superficial actinic porokeratosis, punctate porokeratosis, porokeratosis palmaris et plantaris disseminata and linear porokeratosis.
Linear porokeratosis can be further subdivided in localized, zosteriform, systematized, and generalized. A patient can present more than one type of porokeratosis and any part of the body can be involved.
A 24-year-old woman was referred to our unit with a history of pityriasis rubra pilaris in her lower right leg that had been diagnosed at the age of 6 months. Treatments with corticosteroid and antifungal creams showed no improvement. The lesions were anhidrotic, and present pruritus when exposed to sunlight. Gradually, they expanded to the face, neck, right leg, arm, palm and sole, but remained always restricted to the right hemibody.
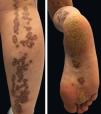
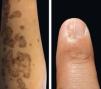
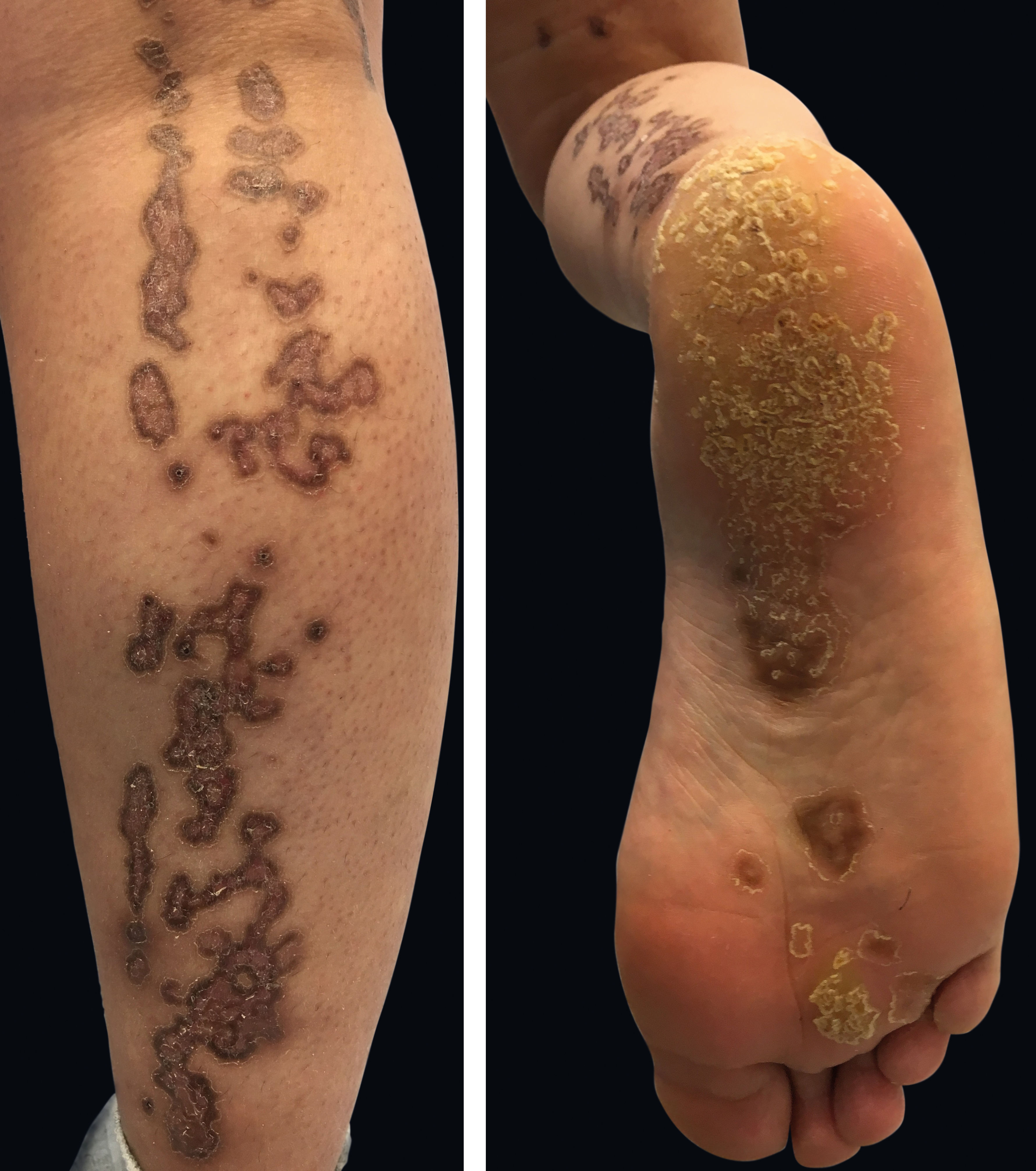
Physical examination revealed deep red to brownish, atrophic plaques with a prominent peripheral hyperkeratotic ridge, coalescing on the lower right leg and the sole, assuming a linear distribution following the Blaschko lines (Figure 1). Similar lesions were present on her face, neck, right arm, and palm and a dorsal pterygium developed on the index finger (Figure 2).
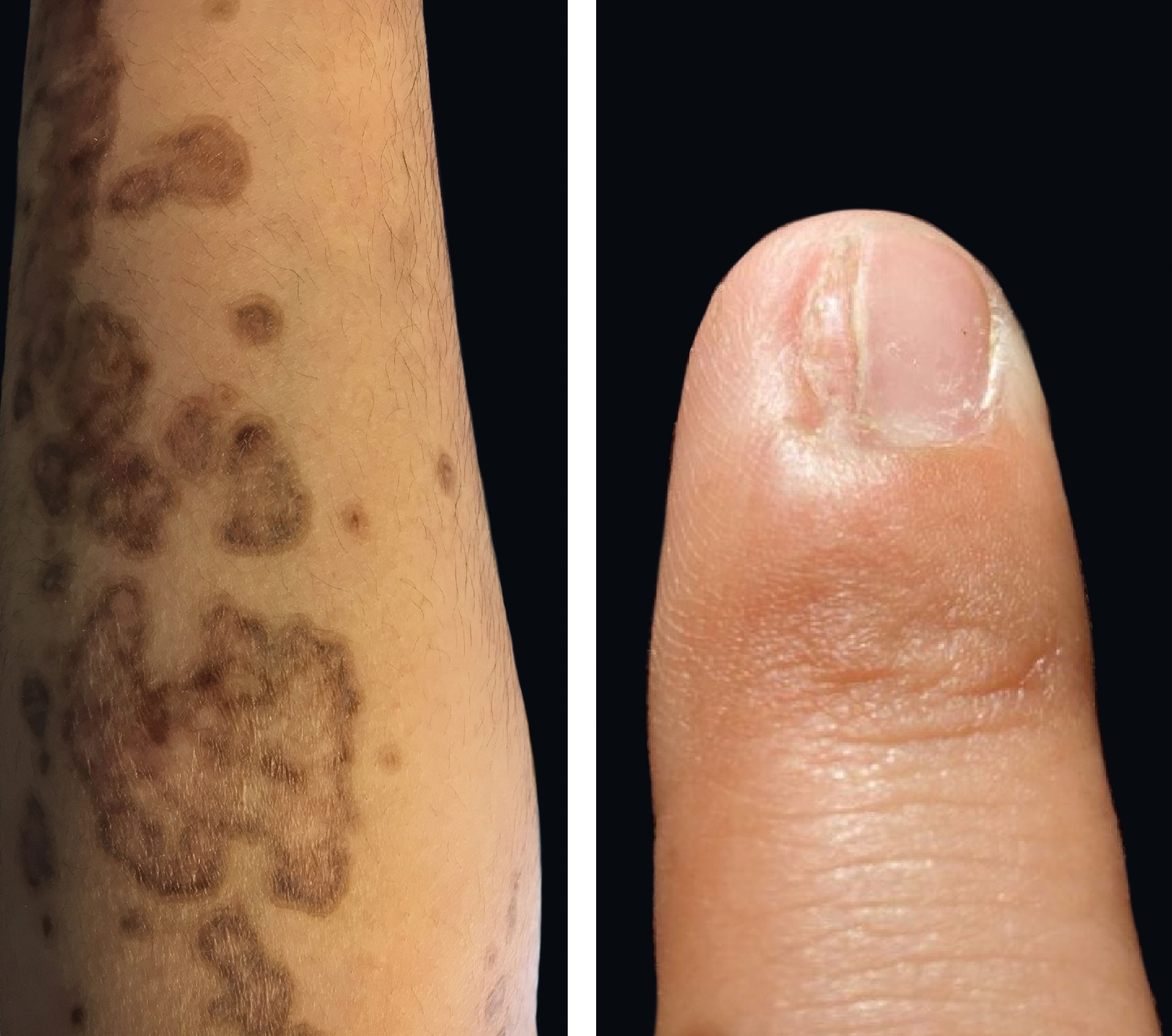
Biopsies of the peripheral ridge and the central atrophic areas revealed the presence of cornoid lamella with loss of the granular layer, and focal lymphocytic infiltrate (Figure 3).
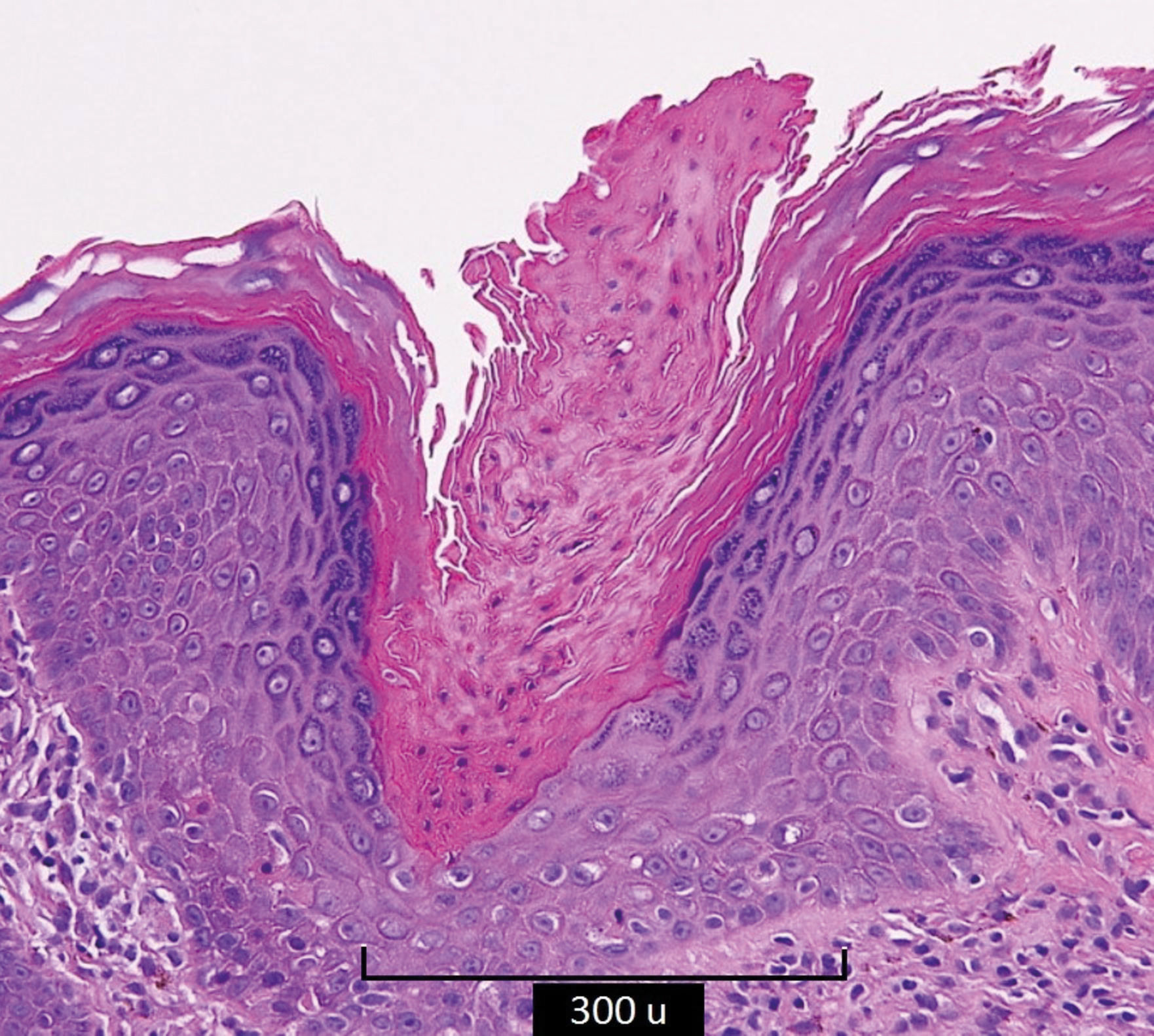
Linear porokeratosis is a strictly unilateral variant with grouped acral lesions and pathology findings identical to those observed in porokeratosis of Mibelli.5 Lesions vary in size, height, and number, may involve the entire hemibody, and slowly grow to form irregular annular plaques with well-demarcated raised borders. The central portion generally presents anhidrosis and alopecia.
Classic porokeratosis of Mibelli, disseminated superficial actinic porokeratosis, porokeratosis palmaris et plantaris disseminata, and punctate porokeratosis are all of autosomal dominant inheritance. Porokeratosis of Mibelli and linear porokeratosis may occur through mosaicism. Reports of coexisting disseminated superficial actinic porokeratosis and linear porokeratosis raise the possibility of a common genetic aberration at chromosomes 12, 15, or 18.2,4,5
Risk factors for porokeratosis include skin burn or damage by ultraviolet radiation A and B, and immunosuppression.2
The cornoid lamella is a constant finding in all variants of porokeratosis. It is formed by a thick column of parakeratotic cells extending outward from a notch in the malpighian layer of the epidermis and forms the raised ridgelike border of the clinical lesion. The cornoid lamella arises in the interfollicular epidermis, extending through the entire corneum stratum. The granular layer is absent and vacuolated keratinocytes can be visualized at the base of the lamella.
Disseminated superficial actinic porokeratosis lesions are small and discrete keratotic papules, with a a less prominent ridge, uniformly distributed on sun-exposed areas.
Porokeratosis palmaris et plantaris disseminata lesions appear on the palms and soles. The keratotic ridge is more pronounced and pruritic. This variant and linear porokeratosis rarely cause bone and/or nail dystrophy.
Punctate porokeratosis lesions are discrete with a thin peripheral ridge.
Diagnosis is clinically confirmed with visualization of a continuous ridge and histopathologic demonstration of the cornoid lamellae.
Most linear porokeratoses present a difficult differential diagnosis with porokeratotic eccrine ostial and dermal duct nevus. The latter presents as multiple linear punctuate pits or keratotic plaques and papules that resemble linear porokeratosis. However, histopathology examination shows epidermal invaginations containing a cornoid lamella that involves the eccrine ducts and hair follicles.
Other differential diagnoses include linear psoriasis, inflammatory linear verrucous epidermal nevus, lichen striatus, incontinentia pigmenti, and pityriasis rubra pilaris.
Up to 20% of the cases with porokeratosis may undergo malignant transformation into basal cell carcinoma or Bowen disease. Fatal outcomes from disseminated squamous cell carcinoma associated with porokeratosis have been reported. Protein p53 has been considered as a possible marker of malignant degeneration in patients with confirmed porokeratosis.2
Treatment includes surgery, cryotherapy, dermabrasion, electrodissection or CO2 laser. Successful treatment has been reported with 5-fluorouracil and imiquimod cream (5%). Retinoid therapy has yielded inconsistent results. A strict patient follow-up is recommended due to risk of malignancy.
