








O ângio‐histiocitoma de células multinucleadas é uma proliferação vascular rara e de caráter benigno, de etiologia ainda desconhecida. Ocorre principalmente em mulheres de meia‐idade e geralmente acomete as regiões acrais; as lesões se apresentam como discretas pápulas violáceas, agrupadas e assintomáticas. Na histopatologia observam‐se proliferação e dilatação de pequenos vasos na derme papilar, estroma fibroso com feixes de colágeno espessados, além de células gigantes multinucleadas. Até o momento, há aproximadamente 140 casos descritos na literatura indexada. É apresentado o caso de uma mulher de 62 anos com quadro clínico típico que, tendo em vista o caráter benigno da doença, optou por não fazer tratamento. São enfatizados os aspectos clínicos e imuno‐histológicos dessa entidade dermatológica incomum.
O ângio‐histiocitoma de células multinucleadas (AHCM) é uma proliferação vascular rara, de caráter benigno, de causa ainda desconhecida. Até o momento, há aproximadamente 140 casos descritos na literatura indexada (PubMed/Medline). É apresentado um novo caso de AHCM com base nos aspectos clínicos e imuno‐histológicos e uma revisão da literatura sobre as principais características clinicopatológicas dessa entidade incomum, frequentemente confundida com dermatofibroma no cenário clínico.
Relato do casoPaciente do sexo feminino, 62 anos, encaminhada devido ao aparecimento de lesões cutâneas no braço esquerdo e no antebraço direito havia oito meses. Não apresentava antecedentes médicos relevantes, negava transplante de órgãos ou uso de medicações imunossupressoras. Os exames laboratoriais gerais não revelaram anormalidades importantes, inclusive com sorologia anti‐HIV negativa. As lesões eram assintomáticas. Nos últimos dois meses, havia notado um novo grupo de lesões no abdômen, na coxa direita e no dorso. Ao exame, foram observadas pápulas eritêmato‐violáceas indolores assimétricas com diâmetro de 3‐10mm (fig. 1).
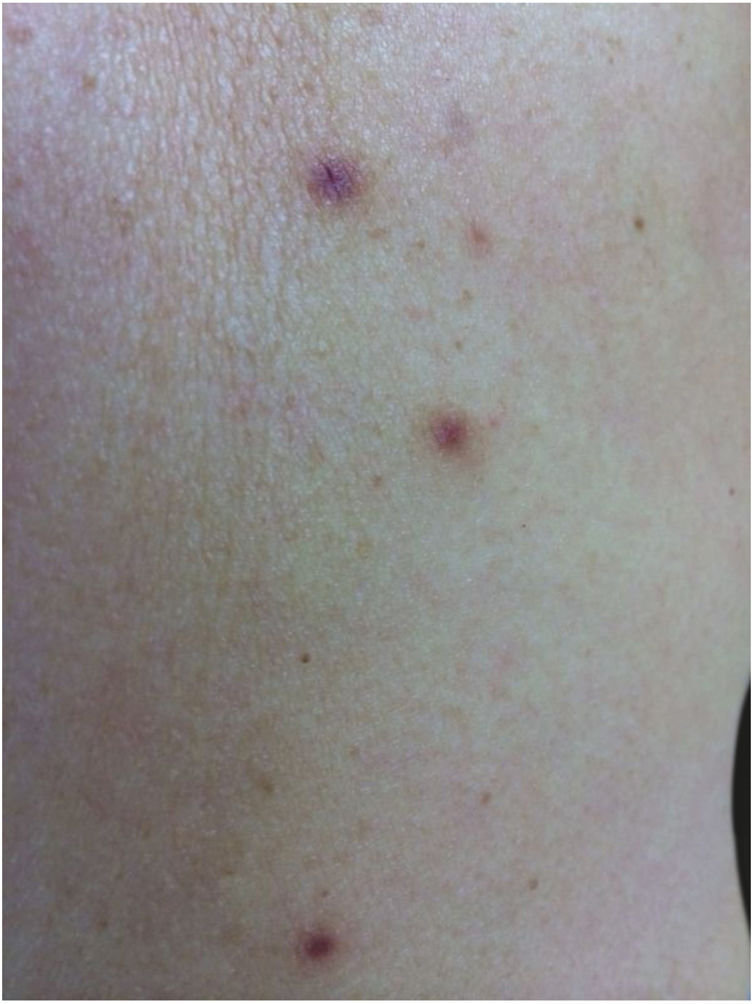
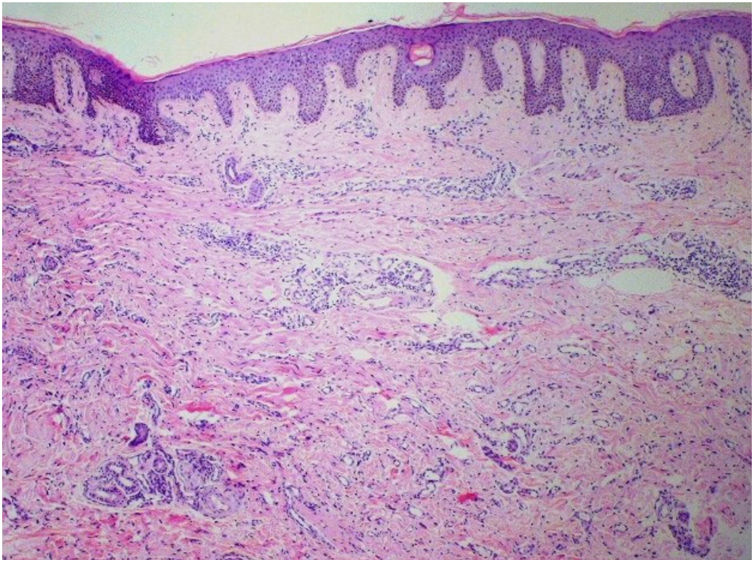
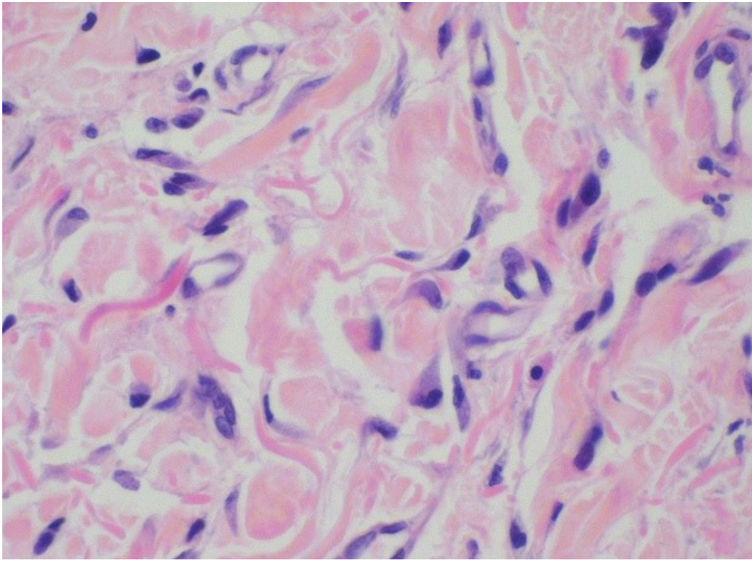
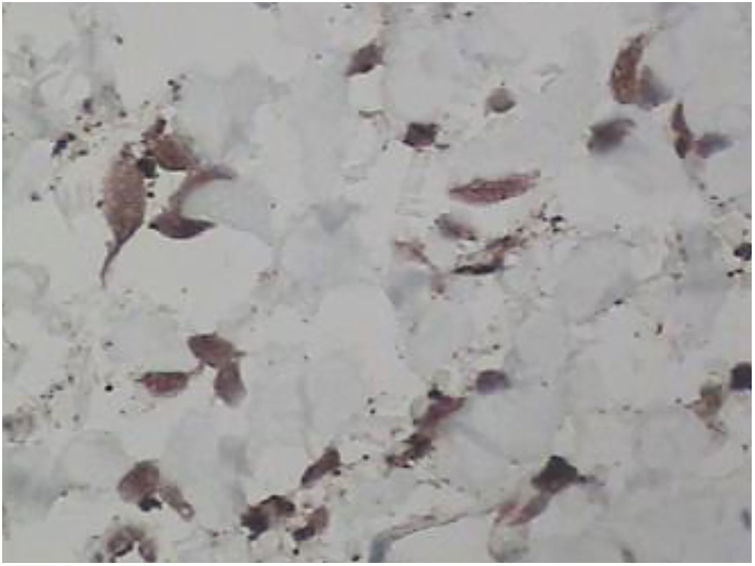
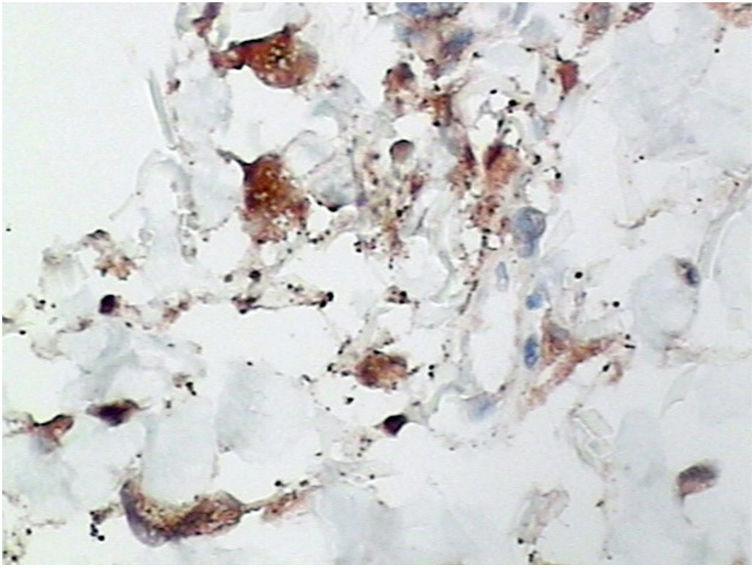
Foi feita biópsia por punch das lesões cutâneas, que mostrou epiderme sem alterações e proliferação fibro‐histiocitária com pequenos vasos na derme reticular, que apresentava células endoteliais proeminentes com resposta inflamatória perivascular composta por células histiocitárias multinucleadas (CM) e alguns plasmócitos. Na derme papilar, foram encontradas proliferação fibroblástica, fibras colágenas espessadas (figs. 2 e 3) e numerosas CM bizarras e irregulares na derme adjacente. O painel de imuno‐histoquímica (IHC) mostrou proteína S‐100 (negativa), FXIIIa (positiva) em CM, CD68 (positiva) em CM, CD34 e CD31 (positiva) em pequenos vasos e CD4 (positiva) em linfócitos dérmicos (figs. 4 e 5). O diagnóstico final foi estabelecido como compatível com AHCM. Pelo caráter benigno da doença, a paciente optou por não fazer tratamento.
A designação AHCM foi originalmente introduzida em 1985 por Smith e Wilson Jones.1 Trata‐se de uma proliferação vascular fibro‐histiocítica rara, de etiologia ainda desconhecida, relatada três vezes mais em mulheres. Comumente acomete indivíduos de meia‐idade (> 40 anos) ou idosos.2,3 Até o presente momento, há cerca de 140 casos de AHCM relatados na literatura indexada (PubMed/Medline). Acredita‐se que sua prevalência seja subestimada devido ao pouco reconhecimento clínico dessa doença.
As lesões são assintomáticas e localizam‐se, na maioria dos casos, nas regiões facial e acral, embora já tenham sido relatadas em outras localizações, como no tronco e, mais raramente, na mucosa.2 Geralmente são unilaterais e apresentam‐se como pápulas avermelhadas, rosadas, violáceas ou acastanhadas de 2 a 15mm de diâmetro, com superfície ligeiramente elevada em forma de cúpula, ou podem ser planas e lisas.2,4 Alguns poucos casos com acometimento bilateral e até mesmo quadro generalizado de AHCM já foram documentados.2 As lesões de AHCM se desenvolvem ao longo de semanas a meses, sem tendência à regressão espontânea.5
Em relação ao diagnóstico clínico diferencial, especial atenção deve‐se dar ao sarcoma de Kaposi, principalmente quando o AHCM se apresenta como pápulas agrupadas.6 Deve‐se incluir também entre os diferenciais: acroangiodermatite, granuloma anular, angiofibroma, dermatofibroma, hemangioma microvenular, líquen plano, linfocitoma e reação à picada de inseto.7
Embora a etiologia e a patogênese do AHCM ainda sejam desconhecidas, entre as hipóteses existentes tem predominado a compreensão de que as lesões sejam resultado de um processo reativo e não neoplásico. Corroboram essa hipótese o fato de ser uma entidade benigna de caráter indolente, a ausência de envolvimento extracutâneo ou transformação maligna e a possibilidade de regressão espontânea, características compatíveis com processo inflamatório reativo em vez de neoplásico.3,8,9
Outra teoria propõe influência hormonal feminina na patogênese, observando a identificação da expressão de receptor de estrógeno alfa (RE) nas células intersticiais e multinucleadas; esse fator explicaria a maior frequência em mulheres. Porém, a identificação de positividade do RE não tem sido consistente em outros casos relatados.3,8
Frew, em 2015, publicou uma revisão com 142 casos de AHCM. O autor levantou a hipótese de que, embora o AHCM tenha origem inicial inflamatória e vascular, os eventos histopatológicos associados à fibrose e à atrofia exercem um papel fundamental na patogênese da doença, especialmente no que se refere à progressão para múltiplas lesões.10
O principal achado histopatológico na AHCM é a proliferação de vênulas e capilares na derme, acompanhada de infiltrado linfocitário e células multinucleadas anguladas. Essas células podem exibir até 10 núcleos hipercromáticos e apresentam citoplasma basofílico; as células expressam vimentina e fator XIIIa, há fibrose dérmica e infiltrado linfo‐histiocitário esparso.2,7,10
Os achados histológicos da MCAH são semelhantes aos de vários tumores benignos cutâneos e outras condições fibroangiomatosas, o que tem levado alguns autores a questionar o status da MCAH como entidade histopatológica independente.6,10 Alguns autores consideram a MCAH como uma variante do dermatofibroma com componente vascular proeminente e células multinucleadas peculiares.10 Outros diagnósticos diferenciais histológicos incluem sarcoma de Kaposi, angiofibroma, linfocitoma cutis, líquen plano e hiperplasia angiolinfoide com eosinofilia.
O diagnóstico diferencial com sarcoma de Kaposi (SK) é importante. Há grande semelhança clínica entre essas condições, porém há algumas diferenças histopatológicas.6,8,10 Microscopicamente, o SK consiste em canais vasculares de anastomose irregular e fendas com aparência de peneira, extravasamento de glóbulos vermelhos, depósitos de hemossiderina e células endoteliais em forma de fuso.3 As células do SK expressam a podoplanina, um marcador do endotélio linfático, que não é expresso pelas células endoteliais do AHCM. A confirmação da positividade do HHV8 (demonstrada com imuno‐histoquímica ou hibridização in situ) permite a distinção entre SK e AHCM.3,10
Na maioria dos casos relatados de AHCM, a IHQ incluiu coloração para fator VIII, fator XIIIa, CD31, CD34, CD68 e vimentina tanto de endotélio vascular quanto nas CM. No artigo de revisão de Frew,10 IHC mostrou 60% das células endoteliais vasculares coradas para CD68. Como esperado, as células endoteliais também expressaram a coloração do fator VIII, CD31 e CD34. No presente caso, os marcadores CD31 e CD34 também foram positivos nessas células. Frew observou que as CM expressavam coloração negativa aos marcadores endoteliais, fator VIII e CD34, com aproximadamente metade dos casos marcando positivamente os macrófagos/histiócitos com fator XIIIa e CD68.10 O fator XIIIa e CD68 também foram marcados na CM deste caso.
Em geral, não é necessário tratamento do AHCM, visto o caráter benigno da condição, a não ser por desejo estético. As lesões podem ser tratadas com corticosteroides intralesionais, excisão cirúrgica, crioterapia, laser de argônio, luz intensa pulsada, laser de CO2. Recentemente, foi publicado um caso tratado com pulsed dye laser com ótimo resultado.3,5
É acrescentado mais um caso de AHCM à literatura, ressaltando‐se a dificuldade de diagnóstico dessa condição na prática dermatológica. É importante estabelecer o correto diagnóstico frente a esse tipo de quadro clínico e, principalmente, para excluir doenças mais graves, como o SK, especialmente no cenário de pacientes com Aids ou transplantados de órgãos.
Suporte financeiroNenhum.
Contribuição dos autoresAnderson Alves Costa: Elaboração e redação do manuscrito.
Glaucia Ferreira Wedy: Concepção e planejamento do estudo.
Walter Belda Junior: Participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados.
Paulo Ricardo Criado: Elaboração e redação do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Conflitos de interesseNenhum.
Como citar este artigo: Costa AA, Wedy GF, Belda Junior W, Criado PR. Multinucleate cell angiohistiocytoma: an uncommon cutaneous tumor. An Bras Dermatol. 2020;95:480–3.
Trabalho realizado no Departamento de Dermatologia, Faculdade de Medicina, Universidade de São Paulo, São Paulo, SP, Brasil.
