








Vitiligo é uma doença complexa cuja patogênese resulta da interação de componentes genéticos, de fatores metabólicos ligados ao estresse oxidativo celular, da adesão dos melanócitos no epitélio e da imunidade (inata e adaptativa), que culminam na agressão aos melanócitos. No vitiligo, os melanócitos apresentam maior sensibilidade ao dano oxidativo, levando ao aumento da expressão de proteínas proinflamatórias como a HSP70. A menor expressão de moléculas de adesão epitelial, como DDR1 e E‐caderina, facilita o dano aos os melanócitos e a exposição de antígenos que favorecem a autoimunidade. A ativação da via do IFN tipo 1 perpetua a ação direta de células CD8+ contra os melanócitos, facilitada pela disfunção de células‐T regulatórias. A identificação de diversos genes envolvidos nesses processos promove o cenário para o desenvolvimento e a manutenção da doença. Entretanto, a relação do vitiligo com fatores ambientais, estresse psicológico, comorbidades e os elementos que definem a suscetibilidade individual à doença são desafios à integração das teorias relacionadas à sua patogênese.
Vitiligo é discromia crônica, adquirida, que promove agressão autoimune contra os melanócitos, resultando em máculas e manchas hipocrômicas ou acrômicas na pele e mucosas, com possível acometimento dos folículos pilosos, em diferentes extensões do tegumento, podendo acompanhar manifestações sistêmicas (p. ex., surdez neurosensorial, uveíte, tireoidite). Sua patogênese é multifatorial; contudo, os mecanismos exatos que integram a suscetibilidade genética do indivíduo, autoagressão aos melanócitos e falha dos mecanismos de tolerância imune, ainda não são completamente elucidados.
A prevalência do vitiligo é bastante variável ao redor do mundo. É mais frequente na África (0,4%), Europa (0,4%) e Oceania (1,2%), que na América do Norte (0,2%) e Ásia (0,1%).1 No Brasil, sua prevalência varia entre 0,46 a 0,68% da população, sem discrepância entre os sexos ou grupos raciais. A idade média de início oscila entre os 20 e 30 anos, apesar de poder acometer desde crianças até idosos. O vitiligo é responsável ainda por 1,4 a 1,9% das consultas dermatológicas, e até 3,5% das consultas dermatológicas em crianças.2,3
Apesar de não apresentar sintomas específicos cutâneos ou implicar em grave risco à saúde, os portadores de vitiligo são impactados em sua qualidade de vida, já que a doença é associada a um forte estigma que compromete as relações sociais, profissionais, autoestima e vestuário; mulheres, adolescentes e pacientes com doenças psiquiátricas são os grupos mais afetados.4
Atualmente, não há uma cura definitiva para o vitiligo; entretanto, vários tratamentos apresentam resultados favoráveis, alcançando algum grau de repigmentação em mais de 80% dos casos.5 O recente avanço na compreensão de sua patogênese promove novas alternativas terapêuticas, que aludem a um futuro mais esperançoso aos pacientes.
Genética do vitiligoO vitiligo é uma doença complexa cujo risco imputado ao componente da herança genética é estimado em 75 a 83%, e os fatores ambientais implicariam nos 20% restantes. Estudos de agrupamento familiar, em gêmeos, e de análises de segregação caracterizam‐no como uma doença multifatorial com padrão de herança poligênica. Em virtude disso, a contribuição individual de cada variante genética para a suscetibilidade é relativamente baixa.6,7
Mapeamento da suscetibilidade do vitiligo por análise de ligaçãoO mapeamento dos fatores de risco genéticos para vitiligo foi realizado por análise de ligação seguida de clonagem posicional. A análise de ligação avalia a segregação não aleatória de regiões cromossômicas entre indivíduos afetados em famílias com vitiligo. Sete loci foram ligados à suscetibilidade ao vitiligo, dos quais quatro foram observados em populações europeias e três em populações chinesas. Um locus no cromossomo 17p13 foi a primeira região genômica ligada ao vitiligo associado a doenças autoimunes.8 A clonagem posicional da região 17p13 identificou o gene NLRP1 como a provável origem do sinal de ligação para vitiligo.9,10
O cromossomo 22q12 foi associado à patogênese do vitiligo.8,11 As variantes que regulam a expressão do gene XBP1 são o fator de risco indicado na região 22q12.12XBP1 é um fator de transcrição que regula a expressão do gene HLA classe II, envolvido na resposta celular ao estresse e frequentemente associado a doenças autoimunes. Em asiáticos e sul‐americanos, foi observada uma ligação entre a região do MHC no cromossomo 6p21 e vitiligo.6,11
Genes relacionados ao HLA de classe I e II foram implicados na patogênese do vitiligo.13,14 O mapeamento fino de HLA por imputação identificou alterações de aminoácidos nos resíduos 135 e 45‐46 para HLA‐DQB1 e HLA‐B, respectivamente, como fortes fatores de risco para vitiligo na população chinesa.15 Além disso, uma variante do promotor que aumenta a expressão de HLA‐A*02: 01 foi associada ao vitiligo comum.16
Três loci em 1p31.3 (p32.2, 7q21.11 e 8p12) foram ligados à suscetibilidade ao vitiligo em europeus, e um em populações asiáticas em 4q13‐q21.17–19 Desses, FOXD3 foi sugerido como o gene causal candidato para 1p31.3 (p32.2) e PDGFRA para 4q13‐q21, enquanto nenhum gene candidato foi sugerido para os dois loci restantes.
Além desses, HLA‐A*33, HLA‐Aw*31, HLA‐DR4, HLA‐DR7 e HLA‐DQB1*0303, dentre outros, são apontados como fatores de risco para o vitiligo em amostras distintas.20,21 Por outro lado, há pesquisas que correlacionam os HLA‐A*09 e HLA‐Aw*19 com menor risco para a doença.21
No Brasil, um estudo com pacientes da região Sudeste demonstrou associação dos HLA‐A*02 e HLA‐DRB1*07 com a suscetibilidade ao vitiligo.20 O HLA‐A*02 também se associa ao risco em populações da China, Índia, Eslováquia e norte da Alemanha. Do mesmo modo, o HLA‐DRB1*07 é identificado em amostras da China, Índia, Eslováquia, Itália, Marrocos, Turquia e Omã.20
Além disso, no Brasil, o HLA‐DQB1*06 é correlacionado à suscetibilidade para o vitiligo (comum, acrofacial e misto), enquanto o HLA‐A*32 para a forma localizada (focal e segmentar). No entanto, esses achados divergem de outras descrições da literatura, sugerindo que os HLA‐DQB1*06 e HLA‐A*32 sejam associados especificamente à população brasileira.20
Genome‐Wide Association Study e a predição de vitiligoOGenome‐Wide Association Study (GWAS) testa um conjunto denso de variantes localizadas em todo o genoma humano para associação com um fenótipo de interesse usando tanto casos‐controles quanto famílias. Até o momento, cinco GWAS foram realizados para vitiligo em populações europeias e asiáticas.22–26 Juntos, eles identificaram mais de 50 loci associados ao risco de vitiligo. A maioria dos loci identificados por GWAS foi detectada em europeus, o que sugere um efeito específico da etnia ou diferenças no desenho ou poder do estudo. No entanto, sete dos loci não MHC foram associados ao vitiligo em indivíduos de diferentes etnias.
A observação dos efeitos do risco em populações independentes fortalece a contribuição global de genes na fisiopatologia do vitiligo. Um desses sete fatores de risco de vitiligo multiétnicos inclui o gene PTPN22, que codifica uma proteína tirosina fosfatase envolvida na sinalização de células T. Curiosamente, variantes em PTPN22 foram associadas a várias doenças relacionadas ao sistema imunológico, incluindo artrite reumatoide, lúpus eritematoso sistêmico e doença de Crohn, que o caracteriza como um gene pleiotrópico para doenças autoimunes.27,28
O gene IKZF4, associado ao vitiligo em europeus e chineses, exerce um silenciamento gênico mediado por FOXP3 em células T regulatórias (Treg).29 O gene FOXP3 localizado no cromossomo X também foi um fator de risco para vitiligo de várias etnias.
Combinadas, essas associações destacam os principais participantes na contribuição das células T para a patogênese do vitiligo. Outros loci candidatos para vitiligo em várias etnias incluem FASLG, um membro da superfamília TNF, e GZMB, uma protease, ambos apontando para um papel de apoptose desregulada no vitiligo. Outra associação não HLA notável com vitiligo encontrada em europeus foi observada para o gene TYR, que regula a biossíntese de melanina em melanócitos.26
O aspecto translacional dos achados de GWAS vai além da descrição dos mecanismos associados à patogênese da doença. A frequência cumulativa dos alelos de risco de vitiligo pode ser usada para calcular a probabilidade, por meio de um escore de risco poligênico, de um indivíduo se tornar um caso. Usando variantes autossômicas de risco de vitiligo descritas por GWAS, o poder preditivo do escore de risco poligênico foi encontrado em 71%, que está entre os maiores valores encontrados para doenças complexas.30
As abordagens dos genes candidatos são conceitualmente baseadas em uma hipótese mecanicista. Foi sugerido que autoimunidade, adesão de melanócitos e disfunção metabólica contribuem para a etiologia do vitiligo, e diversos genes baseados nesses mecanismos já foram testados.31 Exemplos dignos de nota incluem o gene DDR1, que codifica um receptor de tirosina quinase transmembrana que é a principal proteína de adesão dos melanócitos à membrana basal, e se mostrou subregulado no vitiligo, em comparação à pele não afetada.32,33 O DDR1 forma um complexo com a E‐caderina (codificada por CDH1) que é importante para a manutenção da estrutura epitelial. Variantes próximas ao gene CDH1 foram associadas ao vitiligo em brasileiros, relacionando defeitos na adesão de melanócitos à patogênese do vitiligo.34 O dano dos melanócitos em virtude de estresse oxidativo (EO) excessivo é outro mecanismo candidato bem estabelecido para o vitiligo. O acúmulo de espécies reativas de oxigênio (ERO) na epiderme pode inibir a atividade enzimática de BCHE.35 Variantes que controlam a atividade enzimática do gene BCHE foram associadas ao vitiligo em amostras independentes da população brasileira.36
As principais alterações genéticas ligadas ao vitiligo estão resumidas na tabela 1.
Principais genes e antígenos de histocompatibilidade (HLA) envolvidos na patogênese do vitiligo
| Gene | Expressão |
|---|---|
| NLRP1 | + |
| XBP1 | + |
| FOXD3 | + |
| PDGFRA | + |
| PTPN22 | + |
| IKZF4 | + |
| FOXP3 | + |
| DDR1 | ‐ |
| HLA | Risco |
|---|---|
| HLA‐A*02 | ↑ |
| HLA‐Aw*31 | ↑ |
| HLA‐A*32 | ↑ |
| HLA‐A*33 | ↑ |
| HLA‐A*09 | ↓ |
| HLA‐Aw*19 | ↓ |
| HLA‐DQB1*06 | ↑ |
| HLA‐DQB1*0303 | ↑ |
| HLA‐DR4 | ↑ |
| HLA‐DRB1*07 | ↑ |
| HLA‐DR7 | ↑ |
Além da importante redução da melanina e dos melanócitos, a pele com vitiligo apresenta alterações morfológicas tanto no epitélio quanto na derme superior, o que suporta a hipótese que outros elementos contribuam com o desenvolvimento da doença, além da suscetibilidade dos melanócitos ao dano oxidativo e imunológico.
Histologicamente, uma menor pigmentação na camada basal é observada em até 78% das epidermes com vitiligo, e algum infiltrado inflamatório é identificado em até 48% dos casos. Em vitiligos com doença ativa, a histopatologia pode apresentar um padrão de dermatite liquenoide de interface, evidenciando o foco da autoagressão situado na camada basal.37 Linfócitos T (CD3+), especialmente os fenótipos citotóxicos (CD8+), são os predominantes (65,4%) no infiltrado, que é mais evidente na pele perilesional com distribuição perivascular e perianexial. Por esse motivo, a pele perilesional é considerada a área com maior atividade inflamatória no vitiligo (figs. 1 e 2).38
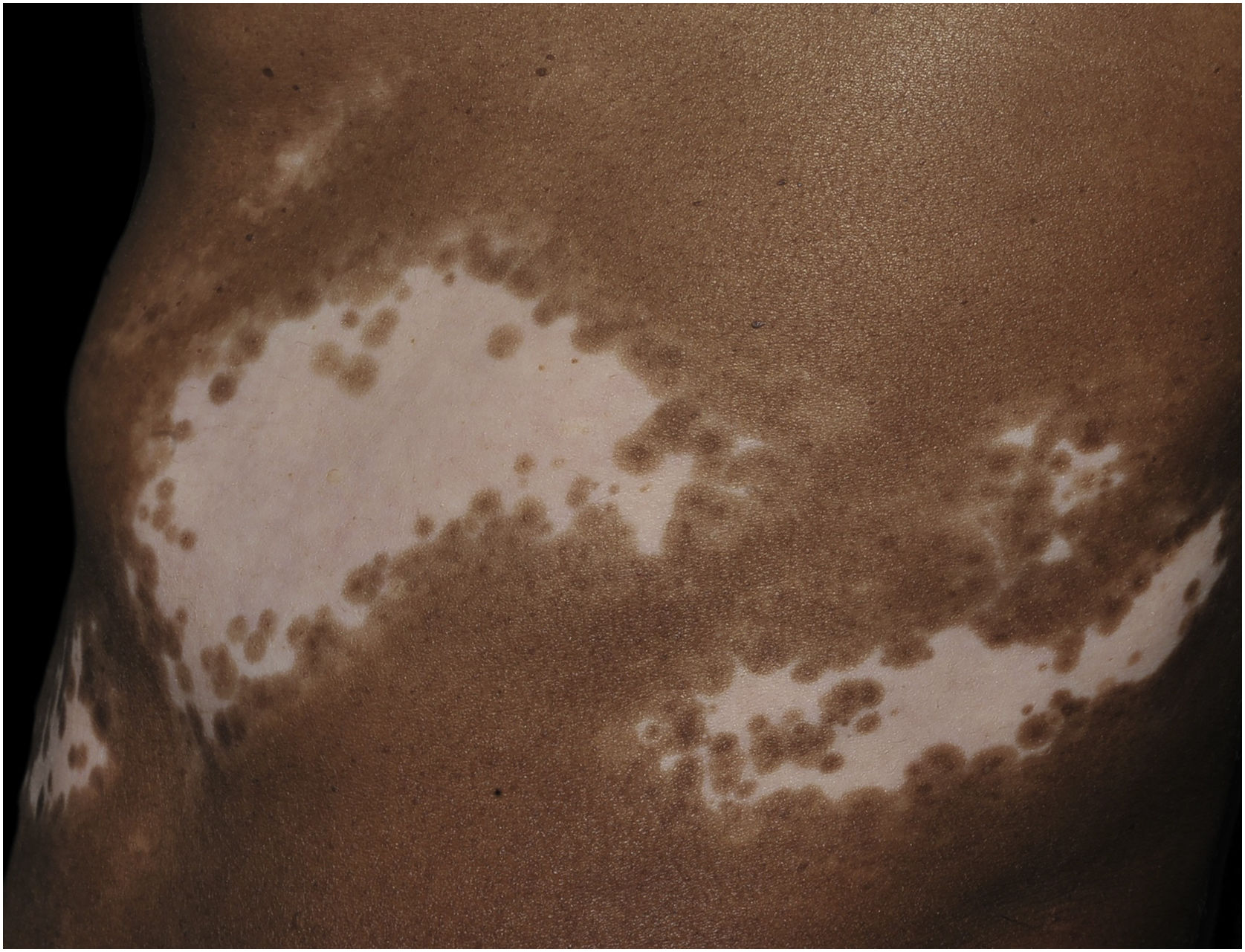
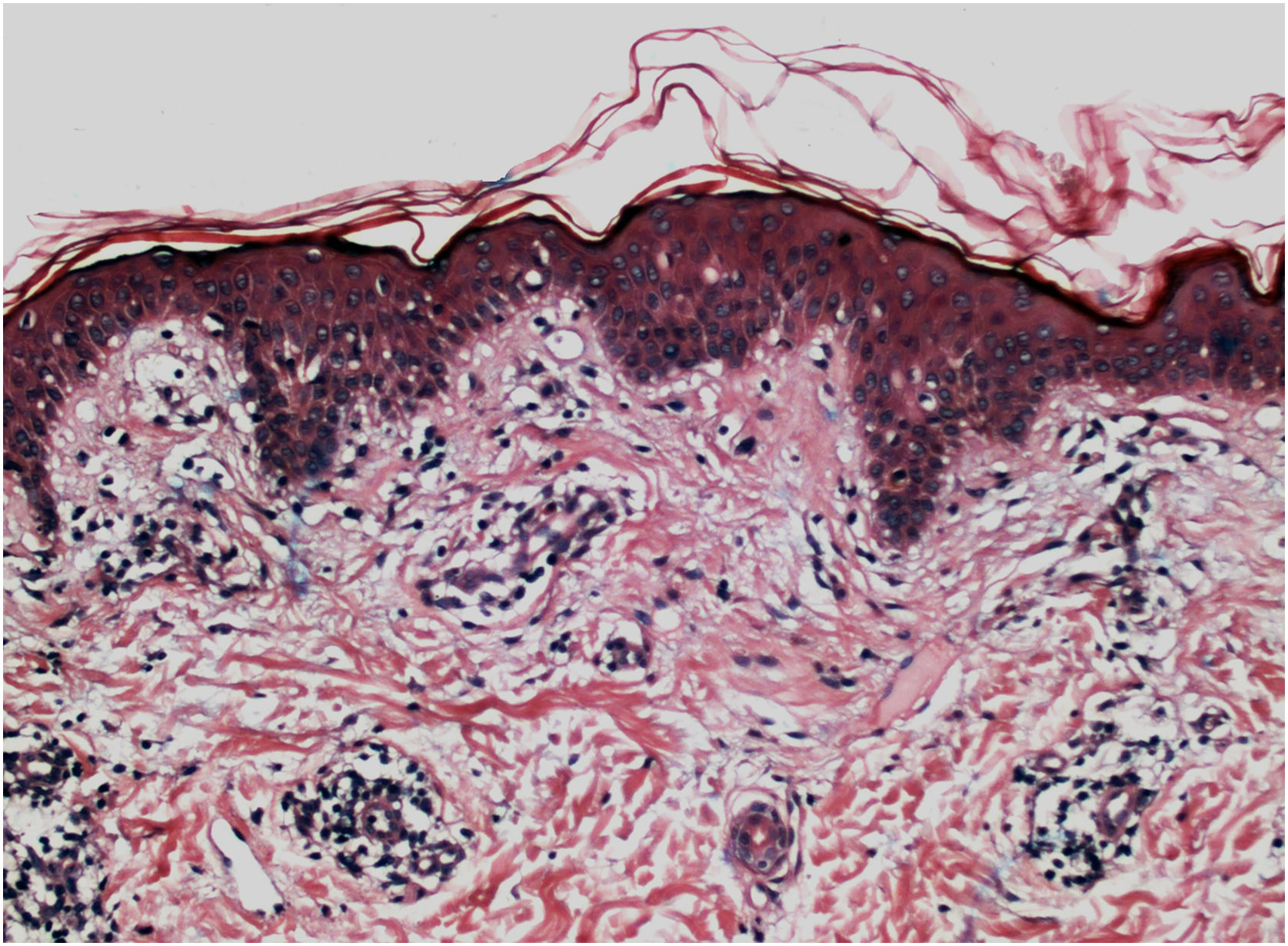
A pesquisa de pigmento melânico pelo Fontana‐Masson identificou melanina residual em 16% dos casos de vitiligo, e, em 12% dos casos, foram identificados melanócitos nas lesões, demonstrando que pode não ocorrer destruição total dos melanócitos de uma área afetada.38
À microscopia eletrônica, identificou‐se melanogênese anormal no vitiligo ativo; os melanócitos da pele controle e da área perilesional de lesões estáveis de vitiligo apresentavam dendritos longos e finos, com número moderado de melanossomos, contrastando com os dendritos dos melanócitos da área perilesional de vitiligo ativo (retráteis e com poucos melanossomos).39 As células de Langerhans encontram‐se aumentadas na epiderme, a membrana basal apresenta‐se espessada, e alterações degenerativas (p. ex., vacuolização citoplasmática) em melanócitos e ceratinócitos da basal são evidenciadas durante a atividade da doença.
O papel dos melanócitos remanescentes na camada basal no processo de repigmentação do vitiligo ainda não é claro, até porque a melanogênese se encontra comprometida nas lesões. Entretanto, a manutenção da pigmentação dos folículos pilosos em uma área afetada representa bom prognóstico, já que a migração de melanócitos a partir da bainha externa folicular pode ser evidenciada pela identificação de repigmentação perifolicular após fototerapia (fig. 1).
Os melanoblastos da bainha externa da raiz do pelo são a principal fonte de repigmentação no vitiligo, quando não atacados pelas células T CD8+. Após estímulo com fototerapia, esses melanoblastos migram, diferenciam‐se em melanócitos e proliferam na direção da epiderme, provavelmente pela regulação positiva do gene associado a células‐tronco, GLI1.40
A camada córnea e a epiderme viável das lesões de vitiligo são aumentadas em espessura, quando comparadas à pele não lesional.41 O epitélio das lesões apresenta ainda corneócitos maiores, supostamente para que compensem a expressão diminuída dos componentes da cornificação e, desse modo, resultam em um estrato córneo mais espesso nas lesões de vitiligo. Uma possível explicação para isso pode ser a expressão reduzida do gangliosídeo D3, que favorece a apoptose ceratinocítica em pacientes com vitiligo. Isso, potencialmente, induz um mecanismo compensatório de espessamento epidérmico para proteger a pele afetada contra a radiação ultravioleta (RUV).
Entretanto, estudos prévios de microscopia óptica e eletrônica demonstraram alterações degenerativas nos ceratinócitos tanto na pele afetada quanto na não afetada de pacientes com vitiligo,42 sugerindo que todo o epitélio desses pacientes apresente suscetibilidade e sofra as pressões patogênicas da doença.
No vitiligo comum há diminuição dos cones epiteliais da junção dermoepidérmica. Do mesmo modo, identificou‐se uma diferença na arquitetura em pacientes com os tipos segmentar e não segmentar de vitiligo. No tipo segmentar existe um notável aumento dos cones epiteliais; já no tipo não segmentar observa‐se acantose, quando comprado com a pele de controles sem vitiligo.37
Defeitos de adesão entre os componentes celulares da epiderme também foram implicados na patogênese do vitiligo. E‐caderina é uma proteína que auxilia na ancoragem entre os ceratinócitos, e sua baixa expressão foi identificada em melanócitos no vitiligo.43 Células positivas para p53 em áreas não lesionais também foram demonstradas na derme, e essa reatividade foi mais elevada em lesões de vitiligo do que em controles.
A redução dos melanócitos no vitiligo também foi relacionada a um defeito de adesão celular, mas não diretamente de apoptose. Além disso, citocinas como IFN‐γ e TNF‐α induzem o destacamento dos melanócitos e diminuem a distribuição de E‐caderina nos melanócitos. Entretanto, a combinação das duas citocinas foi capaz de subregular a expressão do gene CDH1 que codifica E‐caderina, e também reduz a expressão dos genes associados ao déficit de adesão, DDR1 e CCN3. Ainda, MMP‐9 está elevada na pele e no plasma de pacientes com vitiligo comum – é produzida nos ceratinócitos por estímulo de TNF‐α e IFN‐γ e está associada à clivagem da E‐caderina.44
A proteína DKK1, que reduz a melanogênese e certas funções somáticas dos melanócitos, é hiperexpressa por fibroblastos lesionais do vitiligo.45 Da mesma maneira, há maior expressão de fibronectina (proteína central na comunicação intercelular pela ligação com as integrinas) na derme dos pacientes com vitiligo quando comparados com controles sãos.43 Além disso, a elastina é reduzida na derme lesional; no entanto, fibras do colágeno não apresentam diferença quando comparadas com a pele não afetada.46
Alterações oxidativasOs melanócitos cutâneos, localizados no órgão de maior interface com o meio externo e, consequentemente, o mais exposto à RUV e poluentes, são particularmente vulneráveis à produção excessiva de ERO. A concentração dessas substâncias é, inclusive, maior do que em outras células próximas, como ceratinócitos e fibroblastos. Isso se deve, em parte, por sua função especializada de produção de melanina (a qual gera subprodutos como O2− e H2O2) e pela inflamação, decorrentes da fotoexposição excessiva.47,48
No folículo piloso, os melanócitos da bainha externa são responsáveis por intensa síntese de melanina. Há indícios de que a produção de ERO nesses melanócitos favoreça a canície durante o processo de envelhecimento, processo também acompanhado de perda de mecanismos antioxidantes protetores, gerando uma alteração no balanço oxidativo/antioxidativo (status redox). Em um modelo de EO in vitro, a glutamina (aminoácido precursor da molécula antioxidante glutationa) demonstrou ser capaz de reduzir a apoptose de melanócitos. Além de desencadearem a apoptose, as ERO também têm o potencial de reduzir a melanogênese, como demonstrado em testes in vitro após exposição de melanócitos ao H2O2.48–51
A teoria do desenvolvimento de vitiligo em virtude do EO sugere que as ERO teriam sua produção induzida por múltiplos fatores intrínsecos (como a inflamação e a síntese proteica) e extrínsecos, como exposição a RUV, poluentes e derivados fenólicos. Em paralelo, haveria uma falha no mecanismo antioxidante, rompendo a homeostase celular e culminando em dano celular.52
Melanócitos cultivados de áreas não afetadas em pacientes com vitiligo apresentam maior suscetibilidade ao EO que os controles sem vitiligo, evidenciando uma suscetibilidade global do paciente com vitiligo ao dano oxidativo.53
No vitiligo há elevação da enzima superóxido dismutase (responsável por degradar o radical O2− em H2O2 e O2), elevação da peroxidação lipídica (secundária ao EO), além da redução da enzima catalase (conversora de H2O2 em H2O e O2). Entretanto, em comparação com outras doenças inflamatórias, como psoríase e líquen plano, tais alterações também são reportadas, sugerindo que a via do EO não seja doença‐específica. Em contraste, quando é estudada a pele não lesional, há mais alterações na via oxidativa na pele de pacientes com vitiligo do que em outras doenças cutâneas inflamatórias, sugerindo que as áreas despigmentadas sejam parte fenotipicamente alterada de uma pele sujeita ao desequilíbrio óxido‐redutor.54
Além de apresentarem menor capacidade antioxidante sistêmica (redução de glutationa peroxidase) quando comparados a indivíduos sem vitiligo,55 há diferenças na concentração sérica de superóxido dismutase (SOD) e glutationa reduzida (GSH) entre pacientes de vitiligo comum e localizado, pressupondo também uma diferença entre o status sistêmico antioxidante de acordo com a gravidade da doença.56 Polimorfismos no FOXO3A, um gene com importante papel na regulação do EO, também são encontrados em pacientes com vitiligo em atividade.57
O tratamento com UVB de banda estreita, por sua vez, é capaz de equilibrar o status redox em pacientes com vitiligo, demonstrando uma redução significativa nos níveis séricos de malondialdeído (MDA) e aumento de glutationa peroxidase em pacientes tratados por esse método de fototerapia.58
Teoriza‐se que o EO inflija dano celular por meio da indução de apoptose nos melanócitos e inclusive de outros mecanismos de morte celular, como a ferroptose e fagoptose.52 Estudos clínicos e bioquímicos sugerem que, no vitiligo, a superexpressão do gene TRPM2 (transient receptor potential cation channel subfamily M member 2) e do peptídeo CGRP (calcitonin gene‐related peptide) estejam relacionados a canais de cálcio sensíveis ao EO em melanócitos perilesionais. Nesses casos, o H2O2 induziria a desmetilação da região promotora do gene TRPM2 e aumentaria sua expressão nos melanócitos, promovendo o influxo de cálcio para o interior do citoplasma e sua apoptose.59
Em estudo com melanócitos murinos, o processo natural de autofagia, no qual as células degradam suas organelas e proteínas danificadas para manter sua homeostase, também se mostrou prejudicado quando ocorre acúmulo excessivo de ERO.60 No vitiligo, os melanócitos e fibroblastos são mais sensíveis à autofagia induzida por EO, o que reduz seu potencial no controle da melanogênese.61
O EO também pode desencadear alterações no retículo endoplasmático do melanócito, culminando em acúmulo de proteínas defeituosas, estimulando um fenômeno de estresse celular denominado “resposta a proteínas mal enoveladas” (unfolded protein response – UPR).62 Além disso, os requerimentos energéticos para a produção proteica (como a de melanina) por si só geram ERO pelo metabolismo mitocondrial. Essas duas vias parecem estar superativadas em melanócitos de pacientes com vitiligo, o que sugere que tais células toleram menos a demanda de produção de melanina que aquelas de indivíduos saudáveis.63 Mesmo melanócitos saudáveis entram em estresse celular quando expostos a determinados compostos fenólicos como o monobenzil éter de hidroquinona, induzindo a produção de interleucina (IL) 6 e IL8.63,64
Apesar do amplo uso de antioxidantes por pacientes com vitiligo que buscam alternativas terapêuticas, o papel desse mecanismo patogênico e a aplicação desse conhecimento na prática clínica ainda são controversos. A falta de resposta consistente aos antioxidantes sistêmicos sugere que o EO pode não ter uma função central na patogênese da doença, o que gera a necessidade de estudos que identifiquem o real papel do mecanismo nessa doença complexa.54
A figura 3 esquematiza as possíveis vias de dano aos melanócitos em virtude do EO no vitiligo.
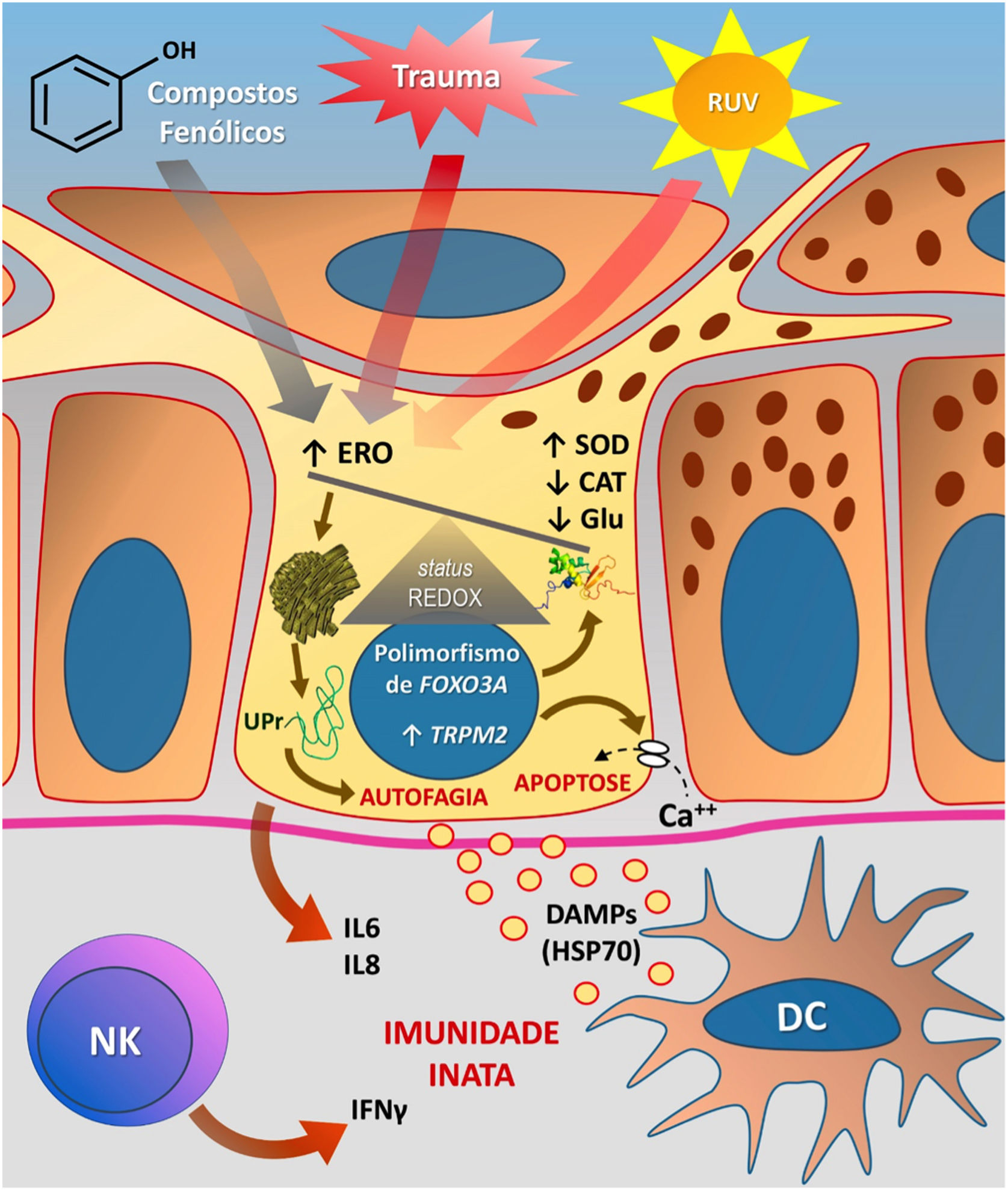
Representação do estresse oxidativo (EO) e da ativação da imunidade inata no vitiligo. As ações da radiação ultravioleta (RUV), compostos fenólicos e trauma (Köbner) aumentam a produção de espécies reativas de oxigênio (ERO). Em paralelo, predisposições genéticas (como mutações no gene FOXO3A) levam a uma ineficiência dos mecanismos antioxidantes, observada pelo aumento da enzima superóxido dismutase (SOD), redução da catalase (CAT) e glutationa (Glu), causando desequilíbrio no status redox. O EO também provoca um acúmulo de proteínas defeituosas no retículo endoplasmático, gerando um fenômeno denominado resposta a proteínas mal enoveladas (unfolded protein – UPr), contribuindo para o processo de autofagia e levando à produção de interleucinas proinflamatórias (IL6 e IL8). O aumento da expressão de TRPM2 (transient receptor potential cation channel subfamily M member 2), também induzido pelo EO, promove influxo de cálcio para o interior do melanócito, culminando com sua apoptose. O EO promove a liberação de padrões moleculares associados a danos (DAMPs), especialmente o HSP70, que iniciam a resposta inata a partir da ativação de células dendríticas (DC) e da participação de células natural killer (NK). Fonte: os autores.
O EO também participa no fenômeno de Köbner, no vitiligo, apesar de sua etiologia ser provavelmente multifatorial. Após o trauma epitelial, mediadores inflamatórios levam ao aumento do EO, induzindo a morte celular, processo também favorecido pelo déficit de adesão dos melanócitos. Além disso, o trauma epitelial promove a liberação de IFN‐α, que aumenta a expressão de CXCL10, favorecendo a migração de linfócitos circulantes.65
Alterações imunológicasImunidade inataAs evidências da participação do sistema imune inato na patogênese do vitiligo são muito consistentes; o vitiligo é considerado o elo entre o EO e a resposta imune adaptativa.66 Primeiramente, células dendríticas, macrófagos e células natural killer (NK) são encontradas na pele lesional e perilesional de pacientes com vitiligo, caracterizando uma ativação da resposta imune inata.67
Células NK ativadas (CD56+/granzima B+) e produtoras de IFN‐γ foram identificadas no sangue e na pele não lesional de pacientes com vitiligo.68 Além disso, há um aumento de citocinas proinflamatórias características da imunidade inata tanto no soro quanto na pele de pacientes com vitiligo, como IL1α, IL1β, IL6, IL8, IL12, IL15 e TNF‐α.69 Esses elementos indicam uma ativação global da imunidade inata do organismo, transcendendo as áreas afetadas.
O EO que ocorre no melanócito, como já mencionado, é possivelmente o gatilho da autoimunidade no vitiligo.63 A comunicação entre o melanócito e o sistema imune inato parece ocorrer pela secreção de exossomos, que contêm antígenos específicos dos melanócitos, miRNAs, proteínas de choque térmico e padrões moleculares associados a danos (DAMPs).64 Esses exossomos entregam autoantígenos às células dendríticas, que sofrem maturação para células apresentadoras de antígenos.70
O DAMP que tem maior evidência de associação com o vitiligo até o momento é a HSP70 (heat shock protein 70).66 A HSP70 é uma proteína da família das chaperonas intracelulares, cuja função é prevenir dobras incorretas de outras proteínas e sua agregação. Enquanto algumas HSPs facilitam o dobramento de proteínas recém‐formadas, outras são particularmente induzidas durante o estresse fisiológico, a fim de gerenciar a carga extra de dobramento incorreto de proteína induzida por estresse, preservando assim a viabilidade da célula.71 Embora as HSPs sejam proteínas intracelulares, já foi observado que, em situações de estresse, elas podem ser secretadas pela célula. No meio extracelular, o reconhecimento dessas proteínas pelas células do sistema imune resulta na transdução do sinal que leva à liberação de citocinas pró‐inflamatórias, como IFN‐α. Estudos in vitro demonstraram a expressão de receptores Lox‐1da HSP70 na superfície das células dendríticas no vitiligo. No mesmo estudo, comprovou‐se que a HSP70 induz a maturação e a ativação das células dendríticas (CD80+).71,72
A HSP70‐induzida está presente na pele lesional e perilesional no vitiligo. Estudos em modelos animais demonstraram a relação entre a HSP70 induzida com o recrutamento e a ativação das células dendríticas inflamatórias no vitiligo. Ainda, em ratos, demonstrou‐se que a HSP70 não só é necessária como é suficiente para acelerar a despigmentação cutânea.73 Por fim, uma pesquisa que introduziu uma HSP70 mutante, na qual foi alterado apenas um aminoácido de sua estrutura, mostrou que ela foi capaz de promover a repigmentação cutânea ao se ligar às células dendríticas e inativá‐las, inibindo, consequentemente, a resposta T‐celular subsequente. Essa molécula de HSP70 com a alteração de um aminoácido é considerada um tratamento potencial para o vitiligo, com a expectativa de ser estudada em ensaios clínicos.74
NLRP‐1 (nuclear localization leucine‐rich‐repeat protein 1) é outro componente da imunidade inata identificado no vitiligo. Variantes da sequência de DNA na região NALP1 estão associadas ao risco de várias doenças autoimunes e autoinflamatórias epidemiologicamente associadas, incluindo o vitiligo comum.9 NLRP‐1 é um regulador chave da resposta imune inata. Ao reconhecer as DAMPs, esse receptor ativa o inflamassomo, que, por meio da via da caspase‐1, induz o processamento da pro‐IL1β em IL1β ativa, assim como sua secreção para o meio extracelular, onde esta acaba por perpetuar a resposta inflamatória.75 A IL1β desempenha um papel fundamental na polarização de células T em direção à Th17.76 A pele perilesional do vitiligo, onde a doença é mais ativa, apresenta aumento da IL1β, sugerindo que essa via também esteja envolvida na progressão do vitiligo.77 Dessa maneira, a inibição da IL1β também pode ser vista como um potencial alvo terapêutico no vitiligo.
Imunidade adaptativaA ativação da imunidade inata desencadeada pelo dano aos melanócitos afetados pelo EO promove a secreção de citocinas e a apresentação de antígenos que resultam na ativação do sistema imune adaptativo, no qual células‐T autorreativas amplificam o dano aos melanócitos na pele afetada pelo vitiligo.
Nesse contexto, os linfócitos T citotóxicos (CD8+) são as principais células implicadas na patogênese da doença.78 Entretanto, apesar de anticorpos reativos contra melanócitos (p. ex., anti‐MelanA, anti‐MCHR1, antitirosinase, anti‐gp100 e antitirosina hidroxilase) apresentarem títulos séricos elevados em pacientes com vitiligo, eles não se correlacionam com a atividade da doença.79 Porém, a literatura é controversa, e o exato papel dos anticorpos antimelanócitos no vitiligo ainda é incerto.
A via do IFN tipo I é um ponto chave para o início da resposta imune do vitiligo. A assinatura de IFN‐I caracteriza um evento precoce e transitório na sua progressão e um elo entre a resposta imune inata e adaptativa.65 Estudos mostram que o IFN‐α, produzido principalmente pelas células dendríticas plasmocitoides (pDC), estimula a produção de quimiocinas, como CXCL9 e CXCL10, por ceratinócitos, levando ao recrutamento de células T com expressão de seu receptor: CXCR3.65,72 Além do mais, a HSP70 contribui potencialmente para a inflamação da pele mediante a ativação de pDC e o aumento da secreção de IFN‐α.72
Células T CD8+ citotóxicas são necessárias e suficientes para a destruição dos melanócitos, atuando como um braço efetor da autoimunidade.78 As lesões são causadas por linfócitos T CD8+ efetores na fase inicial ou ativa da doença e por linfócitos T CD8+ de memória recirculantes e residentes na fase estável. O mecanismo de agressão se baseia na produção de citocinas inflamatórias como TNF‐α e IFN‐γ, além da liberação de granzimas e perforinas que causam dano direto aos melanócitos.78,80 No infiltrado linfocitário da periferia das áreas despigmentadas predominam linfócitos T CD8+, e esse achado se correlaciona com a atividade da doença.81
Antígenos específicos de melanócitos reconhecidos pelas células T CD8+, como MelanA, tirosinase, gp100 e proteínas relacionada à tirosinase 1 e 2, são detectados em maior número no sangue periférico de pacientes com vitiligo quando comparados a controles.82 A pele perilesional também expressa altas concentrações de linfócitos T CD8+ contra antígenos de melanócitos em relação à pele normal.78 Essas células autorreativas são capazes de destruir melanócitos in vitro,81 além de induzir apoptose de ceratinócitos e melanócitos com um padrão semelhante ao da doença.78
O IFN‐γ e os genes que ele induz codificam o receptor de quimiocinas CXCR3 e seus ligantes CXCL9 e CXCL10, fundamentais para a ativação das células T CD8+, encontrando‐se aumentados na pele e no sangue de pacientes com vitiligo.83,84 A CXCL9 promove o recrutamento global de células T autorreativas, mas sem ação efetora, enquanto a CXCL10 é necessária para a progressão e a manutenção da doença.85 Além disso, a neutralização de CXCL10 em modelos animais previne o surgimento de novas lesões e induz a repigmentação das áreas já estabelecidas,85 identificando o potencial terapêutico da inibição de eixo IFN‐γ/CXCL10/CXCR3.86 Os ceratinócitos são as principais fontes dessas citocinas, e a dosagem de CXCL9 e CXCL10 é um potencial biomarcador de atividade da doença.84
A via Janus kinase/transdutores de sinal e ativadores de transcrição (JAK/STAT) participa da imunopatogênese do vitiligo por meio de sua interação com IFN‐γ. A sinalização de IFN‐γ/CXCL10 inicia com a ligação de IFN‐γ a seu receptor heterodimérico, que estimula a via JAK/STAT e leva à ativação de STAT1. Em seguida, ocorre a translocação de STAT1 para o núcleo e subsequente ligação à região promotora de genes induzidos pelo IFN‐γ, como CXCL10. Há quatro membros da família de proteínas JAK, incluindo JAK1, JAK2, JAK3 e tirosina quinase 2 (TYK2). JAK1 e a JAK2 estão diretamente envolvidas na sinalização de IFN‐γ. Nesse contexto, há pesquisas usando diversos inibidores de JAK como uma estratégia para a repigmentação.87
A recidiva do vitiligo é um fenômeno comum, ocorrendo frequentemente no mesmo local envolvido previamente ao tratamento. Esse padrão de recorrência sugere o papel da memória autoimune na manutenção do quadro, caracterizada pela presença de células T residentes de memória (TRM) CD8+ tanto na epiderme quanto na derme, que agem como sentinelas para o recrutamento de células T de memória da circulação.80 As TRM são linfócitos de longa duração que se desenvolvem após o início de uma resposta imune mediada por células T.
Com base na demonstração de que as TRM necessitam de IL15 para sua diferenciação, a interrupção da via IL15 usando um anticorpo anti‐CD122 ocasionou reversão da despigmentação em camundongos com vitiligo estável. Nesse ensaio, o tratamento de curto prazo com anti‐CD122 inibiu produção de IFN‐γ pelas TRM, e o uso a longo prazo depletou essas células das lesões.88 Funções adicionais da IL15 também foram mapeadas em outros trabalhos. Um estudo evidenciou que a IL15 provoca a expressão de NKG2D em células T CD8+ efetoras de memória na pele com vitiligo, suscitando a produção de IFN‐γ e de TNF‐α.89 Além disso, foi demonstrado que o EO induz a transapresentação de IL15 nos ceratinócitos, contribuindo para a ativação de células T CD8+ efetoras de memória mediante a ativação da via JAK/STAT (JAK1‐STAT3 e JAK3‐STAT5).90 Desse modo, o bloqueio da sinalização da IL15 parece ser promissor na busca de novos tratamentos.
A figura 4 detalha a transapresentação de IL15 nos ceratinócitos induzida pelo EO e a interação do IFNγ com a via JAK/STAT.
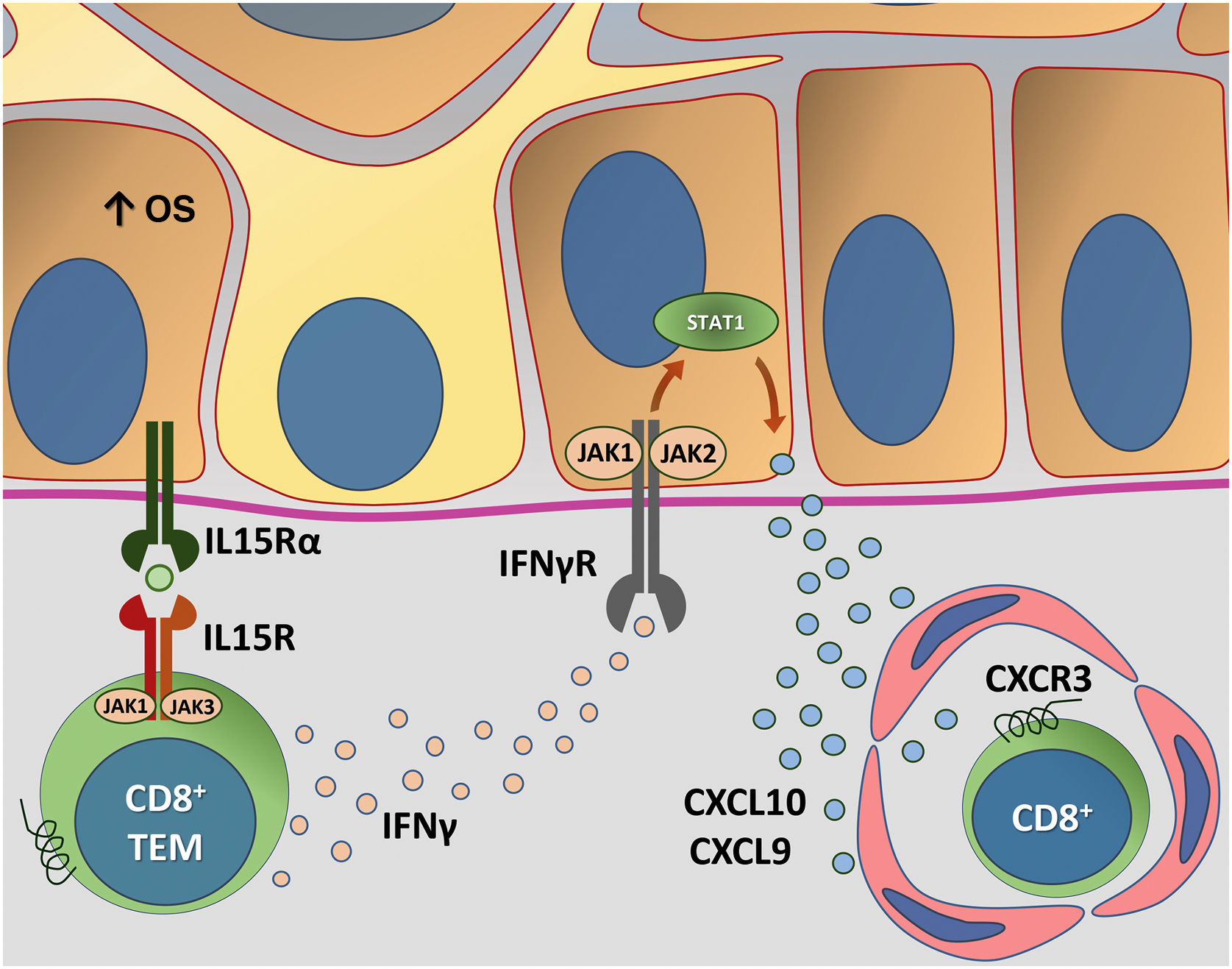
Representação da transapresentação de interleucina (IL) 15 nos ceratinócitos induzida pelo estresse oxidativo (EO) e da interação do interferon gama (IFN‐γ) com a via Janus quinase/transdutores de sinal e ativadores de transcrição (JAK/STAT). O EO promove a transapresentação de IL15 nos ceratinócitos por meio da ligação de IL15 ao receptor heterodimérico de IL15 (IL15R) nos linfócitos T CD8+ efetores de memória (TEM), composto por CD122 e CD132, e ao receptor de IL15α (IL15Rα) nos ceratinócitos (CD215). Esse processo potencializa a ativação de células T CD8+ efetoras de memória e a produção de citocinas inflamatórias, como IFN‐γ, via sinalização JAK/STAT (JAK1‐STAT3 e JAK3‐STAT5). O eixo IFN‐γ/STAT1/CXCL10 conduz a destruição autoimune dos melanóctios. O IFN‐γ sinaliza por meio do receptor de IFN‐γ (IFN‐γR) para estimular JAK1/JAK2 e ativar STAT1. A ativação induz a produção de CXCL9 e CXCL10, que sinaliza por meio do receptor CXCR3 para o recrutamento de mais células T CD8+ autorreativas. Fonte: os autores.
As células Tregs CD4+ atuam na manutenção da tolerância aos próprios tecidos por meio da supressão da atividade das células T efetoras. No vitiligo, há disfunção de Tregs, embora não se saiba exatamente se por inabilidade de migrar para a pele, diminuição do número ou supressão da atividade.91 Em modelos animais de vitiligo, há controle da progressão da doença e repigmentação das lesões com o reestabelecimento da população de Tregs.92
Alterações do sistema imune adaptativo são evidenciadas tanto no vitiligo segmentar quanto no não segmentar, apesar de diferentes padrões de reatividade. No vitiligo não segmentar ocorre ativação imune sistêmica, enquanto no segmentar há apenas reação citotóxica localizada, provavelmente em virtude do mosaicismo dos melanócitos.93 Tregs não são afetadas no sangue periférico do vitiligo segmentar, em comparação com controles. Por outro lado, Tregs estão reduzidas no vitiligo não segmentar e associadas a outras comorbidades autoimunes, menos frequentes no vitiligo segmentar. Além disso, a resposta de anticorpos contra antígenos específicos de melanócitos ocorre apenas no vitiligo não segmentar.
A maior frequência de comorbidades autoimunes em pacientes com vitiligo não segmentar e também em seus parentes reforça a ideia do vitiligo como uma representação fenotípica de um desequilíbrio autoimune sistêmico. As comorbidades variam amplamente com a população estudada, e dependem do gênero, idade, raça e subtipo do vitiligo. A tabela 2 lista as principais doenças autoimunes e autoinflamatórias descritas no vitiligo; as tireoidites autoimunes são as mais frequentes e bem estabelecidas. Destacam‐se, ainda, alterações oculares e auditivas decorrentes da presença de melanócitos na úvea e na cóclea.3,94
Principais doenças autoimunes e autoinflamatórias associadas ao vitiligo
| Doença autoimune ou autoinflamatória |
|---|
| Alopecia areata |
| Anemia perniciosa |
| Artrite reumatoide |
| Dermatite atópica |
| Dermatomiosite |
| Diabetes mellitus tipo 1 |
| Doença de Addison |
| Doença de Crohn |
| Doença de Graves |
| Esclerodermia sistêmica |
| Esclerose múltipla |
| Lúpus eritematoso sistêmico |
| Psoríase |
| Retocolite ulcerativa |
| Síndrome de Sjögren |
| Tireoidite de Hashimoto |
O desenvolvimento de vitiligo e halo nevus em pacientes com melanoma é um fenômeno bem descrito, provavelmente em virtude da autoagressão aos melanócitos induzida por antígenos tumorais. Entretanto, isso ocorre entre 10 e 25% dos pacientes tratados com inibidores de checkpoint (p. ex., nivolumabe, pembrolizumabe), uma nova classe de medicação para melanoma metastático. Essas lesões clínicas são indistinguíveis do vitiligo comum, e o fenômeno é chamado de leucoderma ou vitiligo‐like, porém, há dúvida se sua origem decorreria da citotoxicidade direta aos melanócitos ou do desenvolvimento de autoimunidade (vitiligo) em pacientes predispostos.95
A figura 5 esquematiza as principais alterações da imunidade adaptativa envolvidas no vitiligo.
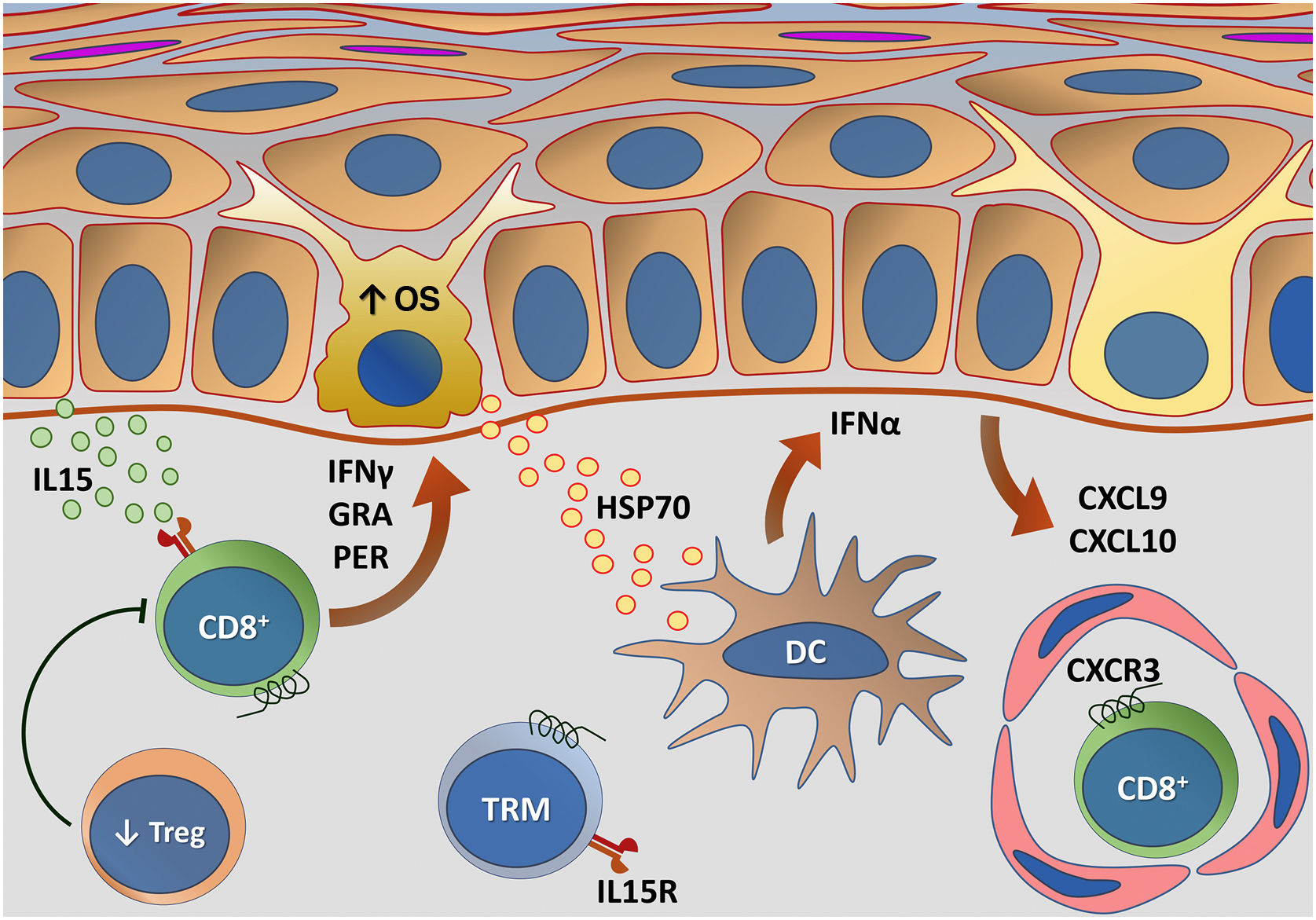
Representação das alterações relacionadas com a imunidade adaptativa no vitiligo. Os melanócitos afetados pelo estresse oxidativo (EO) provocam a ativação da imunidade inata por meio da secreção de exossomos, que contêm padrões moleculares associados a danos (DAMPs), especialmente a heat shock protein 70 (HSP70). A HSP70 estimula a secreção de IFN‐α pelas células dendríticas na fase inicial da progressão da doença, que induz a produção das quimiocinas CXCL9 e CXCL10 pelos ceratinócitos e o recrutamento de células T com expressão do receptor CXCR3. CXCL10 apresenta ação efetora, enquanto CXCL9 atua no recrutamento global de células T CD8+ autorreativas. Células T CD8+ efetoras são responsáveis pela destruição dos melanócitos mediante a produção de interferon gama (IFN‐γ), liberação de granzimas e perforinas, facilitada pela disfunção de células T regulatórias (Treg). As células T residentes de memória (TRM) CD8+ se desenvolvem após o início da resposta imune mediada por células T e estão implicadas na manutenção da doença, sendo retidas no tecido em virtude da transapresentação de IL15 pelos ceratinócitos. Fonte: os autores.
O exato mecanismo pelo qual as terapias tópicas com corticoides e inibidores de calcineurina interferem na patogênese da doença ainda não foi completamente elucidado, mas acredita‐se que reduzam o infiltrado linfocitário e a expressão de TNF‐α. Em lesões induzidas por trauma (Köbner), o tratamento com corticoide tópico e tacrolimus reduziram o infiltrado inflamatório subjacente.96
A fototerapia, além de estabilizar o equilíbrio redox, age suprimindo a resposta inflamatória e promovendo a apoptose de células‐T, a diminuição de citocinas inflamatórias, o aumento de IL10 (com indução de células Tregs), a redução da expressão de JAK1 e a depleção de células de Langerhans da epiderme.97
Vitiligo induzido por agentes químicosO vitiligo induzido por produtos químicos, também chamado leucoderma, foi descrito especialmente em trabalhadores da indústria da borracha, que têm contato com derivados fenólicos. O agente primeiramente identificado foi o monobenzil éter de hidroquinona (monobenzona), que induz a morte celular sem ativação da cascata da caspase ou fragmentação do DNA.98
Outro composto fenólico usado em clareadores cutâneos foi responsável por um surto de leucoderma no Japão. O rododendrol promove citotoxicidade via um mecanismo dependente da tirosinase.99
Tinturas de cabelos também foram implicadas no aparecimento de leucoderma; um dos principais suspeitos é a parafenilenodiamina. Todavia, o mecanismo causal pelo qual derivados do benzeno causam despigmentação ainda não foi elucidado.100
Leucodermas químicos, porém, não compartilham as alterações de autoimunidade (p. ex., anticorpo anti‐TYRP1), como ocorre no vitiligo, apesar de promoverem dano direto aos melanócitos.101
Teoria neuralA teoria neural do vitiligo baseia‐se na observação clínica da forma de distribuição das lesões: sobre dermátomos no tipo segmentar, e simétrica no vitiligo não segmentar, aludindo à influência neuroimunológica no vitiligo. Há ainda casos anedóticos de surgimento de vitiligo em áreas delimitadas após lesão neural.102 Finalmente, o estresse psicológico extremo é um fator classicamente associado ao desenvolvimento de vitiligo e de outras doenças autoimunes, embora o mecanismo fisiopatológico implicado nesse contexto não esteja completamente elucidado, se dependente da mudança no estado oxidativo, da modificação do sistema de tolerância imune ou da participação de neurocininas.103
No vitiligo, o neuropeptídeo Y encontra‐se aumentado na pele lesional e na área perilesional.104 Já a substância P apresenta‐se aumentada na pele do vitiligo estável quando comparado à doença instável, sugerindo associação com a estabilidade do vitiligo.105
O estresse físico ou psicológico extremo aumenta a síntese de catecolaminas e demanda a ativação do eixo hipotálamo‐hipófise‐adrenal, que interfere na regulação do sistema imune. Na fase aguda do estresse há neutrofilia e aumento das células NK no plasma, além do desbalanço das citocinas proinflamatórias, com aumento da IL6 em decorrência da secreção de cortisol e de catecolaminas, modulando a resposta imune e o desenvolvimento de doenças autoimunes e autoinflamatórias.106
Terapias emergentesAs três classes terapêuticas emergentes do vitiligo que estão em estado mais adiantado de desenvolvimento e que são associadas às vias patogênicas mais importantes são os inibidores de tirosina quinases (p. ex., JAK1, JAK2, JAK3, TYK2), anti‐IL15 e HSP70 mutante.74,88,107 A tabela 3 apresenta os medicamentos emergentes, de acordo com seu alvo fisiopatológico.
Principais terapias emergentes e alvos terapêuticos no vitiligo
| Classe/Fármaco | Alvo terapêutico/mecanismo de ação |
|---|---|
| Proteína HSP70 mutante | Inibição da imunidade inata ‐ redução da ativação de células dendríticas |
| HSP70iQ435A | |
| Inibidores de tirosina quinases | Imunidade adaptativa ‐ alteração na sinalização de citocinas/quimiocinas (IFNy, CXCL10, IL15) |
| Tofacitinibe (JAK1/3) | |
| Ruxolitinibe (JAK1/2) | |
| Cerdulatinibe (SYK/JAK 1/2/3) | |
| ATI‐50002 (JAK1/3) | |
| PF‐06651600 (JAK3) | |
| PF‐06700841 (TYK2/JAK1) | |
| Biológicos anti‐IL15 | Imunidade adaptativa ‐ bloqueio da sinalização de IL15 |
| Anti‐CD122 mAb | |
| AMG 714 (anti‐IL15 mAb) | |
HSP70, heat shock protein 70; JAK, Janus quinase; SYK, tirosina quinase do baço; TYK, tirosina quinase; IFN‐γ, interferon gama; IL, interleucina; mAb, anticorpo monoclonal.
A compreensão da patogênese do vitiligo evolui em paralelo ao conhecimento da regulação genética dos fenômenos ligados ao EO e à resposta imune cutânea.
O controle epigenético da expressão de genes ligados a esses fenômenos pode ser um integrador desses processos, já que fatores ambientais, dietéticos, padrões de resposta comportamental ao estresse e da crescente exposição a compostos industrializados são intensamente presentes na vida moderna, principalmente a poluição do ar e compostos aromáticos.108,109
A regulação das vias relacionadas às vesículas de transporte (exossomos) entre ceratinócitos, melanócitos e as células inflamatórias constitui outro fator que deve elucidar a interação entre o processo de dano celular, melanogênese e resposta imune no vitiligo.110
O papel da disbiose intestinal na modulação da inflamação, EO e autoimunidade pode revelar perfis de suscetibilidade e expressão clínica do vitiligo, já que maior permeabilidade intestinal favorece a ativação da imunidade inata e promove estímulo inflamatório sistêmico.111
Por fim, a importância da hipovitaminose D, uma epidemia moderna, na regulação da resposta imune, assim como o efeito de sua suplementação como coadjuvante ao tratamento, precisam ser explorados, uma vez que pacientes com vitiligo apresentam níveis de vitamina D mais baixos.112
Em conclusão, a patogênese do vitiligo resulta de uma interação de elementos (multifatorial) que pondera uma base de suscetibilidade genética sobre a qual incidem fatores ambientais, EO, fatores psicológicos e padrões de resposta autoimune. A integração dessas teorias é o principal desafio na construção de modelos fisiopatológicos.
Suporte financeiroNenhum.
Contribuição dos autoresHelena Zenedin Marchioro: Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; revisão crítica da literatura; revisão crítica do manuscrito.
Caio César Silva de Castro: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; revisão crítica da literatura; revisão crítica do manuscrito.
Vinicius Medeiros Fava: Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; revisão crítica da literatura; revisão crítica do manuscrito.
Paula Hitomi Sakiyama: Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; revisão crítica da literatura; revisão crítica do manuscrito.
Gerson Dellatorre: Aprovação da versão final do manuscrito; elaboração e redação do manuscrito; revisão crítica da literatura; revisão crítica do manuscrito.
Hélio Amante Miot: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; revisão crítica da literatura; revisão crítica do manuscrito.
Conflito de interessesCaio César Silva de Castro: Advisory Board – Abbvie, Sun Pharma e Aché. Hélio Amante Miot: Advisory Board – ĹOréal; Merz.
Como citar este artigo: Marchioro HZ, Silva de Castro CC, Fava VM, Sakiyama PH, Dellatorre G, Miot HA. Update on the pathogenesis of vitiligo. An Bras Dermatol. 2022;97:478–90.
Trabalho realizado no Departamento de Dermatologia, Faculdade de Medicina, Universidade Estadual Paulista, Botucatu, SP, Brasil.
