
















A epidermólise bolhosa hereditária compreende um grupo de doenças genéticas caracterizadas por fragilidade cutânea e formação de bolhas na pele e nas mucosas em resposta a trauma mínimo. A epidermólise bolhosa é clínica e geneticamente muito heterogênea, classificada em quatro tipos principais de acordo com a camada da pele na qual ocorre a formação de bolhas: simples (intraepidérmica), juncional (dentro da lâmina lúcida da membrana basal), distrófica (abaixo da membrana basal) e de Kindler (padrão misto de clivagem da pele). Além disso, a epidermólise bolhosa é subclassificada em diversos subtipos, que consideram as características clínicas, a distribuição das bolhas e a gravidade dos sinais cutâneos e extracutâneos. Variantes patogênicas em pelo menos 16 genes, que codificam proteínas essenciais para integridade e adesão das camadas da pele, já foram associadas aos distintos subtipos de epidermólise bolhosa. A marcante heterogeneidade da doença, que inclui fenótipos com amplo espectro de gravidade e muitos genes causais, dificulta sua classificação e seu diagnóstico. Por isso, dermatologistas e geneticistas reúnem‐se periodicamente a fim de revisar e atualizar os critérios de classificação da doença. A presente revisão tem como objetivo atualizar o estado da arte sobre a epidermólise bolhosa hereditária, com foco especial nos aspectos clínicos e genéticos associados, e trazer dados do mais recente consenso de reclassificação, publicado em 2020.
A epidermólise bolhosa hereditária (EB) compreende um grupo de doenças genéticas associadas à fragilidade cutânea, que leva à formação de bolhas, erosões e cicatrizes na pele e em mucosas em resposta a mínimos traumas mecânicos.1 A EB é clínica e geneticamente muito heterogênea, compreende fenótipos com níveis contrastantes de gravidade e envolve alterações em pelo menos 16 genes.2 Dada tal heterogeneidade, a EB é classificada em quatro tipos principais e, ainda, em diversos subtipos clínicos. A classificação principal está relacionada com a camada da pele na qual ocorre a formação de bolhas: EB simples (EBS; camada intraepidérmica), EB juncional (EBJ; dentro da lâmina lúcida da membrana basal), EB distrófica (EBD; abaixo da membrana basal) e EB de Kindler (EBK; padrão misto de clivagem da pele).1–3
Outras desordens relacionadas à fragilidade da pele são classificadas em categorias separadas, seja porque a formação de bolhas é uma parte mínima do quadro clínico ou porque a clivagem da pele se dá de maneira muito superficial.2 Entre elas, destacam‐se as desordens de descamação da pele, as erosivas, as hiperceratósicas e as do tecido conectivo com fragilidade da pele.2 Até a mais recente reclassificação dos tipos de EB, publicada em 2020, muitas dessas desordens eram consideradas subtipos de EB.2,3
Os dados epidemiológicos acerca da incidência e prevalência da EB no mundo são bastante variáveis entre os estudos. Um dos principais grupos de pesquisa em EB, em uma amostra de 3.271 pacientes com EB dos Estados Unidos, estimou a incidência (entre 1986‐2002) e a prevalência (2002) da doença no país em 19,57 e 11,07 por um milhão de indivíduos, respectivamente.4 No Brasil não há estudos epidemiológicos sobre a EB; entretanto, a Debra‐Brasil (associação de apoio a pacientes com EB do Brasil) apresenta registro de mais de 900 pacientes no país (dados não publicados).
Os genes associados à EB codificam proteínas com funções estruturais na epiderme, na zona da membrana basal ou na porção superior da derme e que são importantes para a integridade da pele e para a adesão derme‐epiderme.5 Desse modo, alterações genéticas que alteram a dinâmica e a função dessas proteínas resultam em falha das estruturas que conferem estabilidade mecânica à epiderme (como o citoesqueleto de queratina e os desmossomos) e à zona da membrana basal (como os hemidesmossomos, as adesões focais, os filamentos de ancoragem e as fibrilas de ancoragem) (fig. 1).6 A camada na qual as bolhas se desenvolvem nos diferentes tipos de EB está correlacionada à localização da proteína alterada na estrutura da pele.

Representação das camadas da pele associadas aos diferentes tipos de epidermólise bolhosa (EB). Na epiderme, os queratinócitos estão retratados. A epiderme é anexada à derme pela membrana basal, formada pela lâmina lúcida e lâmina densa. Ao lado esquerdo da figura é apresentado cada tipo principal de EB próximo à respectiva camada da pele na qual ocorre a formação das bolhas. A clivagem na EB simples (EBS) ocorre dentro dos queratinócitos basais; na EB juncional (EBJ), dentro da lâmina lúcida; e na EB distrófica (EBD), na sublâmina densa, na porção superior da derme (derme papilar). Na EB de Kindler (EBK), a clivagem pode ocorrer nos queratinócitos basais, na lâmina lúcida ou abaixo da lâmina densa. Ao lado direito da figura são apresentados os principais complexos de adesão das camadas da pele que estão associados aos subtipos de EB. Em destaque estão representados os hemidesmossomos, os filamentos de ancoragem e as fibrilas de ancoragem, que têm papel essencial para a estável adesão dos queratinócitos basais da epiderme à zona da membrana basal e para a ligação da lâmina densa à porção superior da derme.
O fenótipo característico das diferentes formas da EB está correlacionado ao gene que está alterado. Por outro lado, diferentes genes também levam a fenótipos muito similares de EB, o que adiciona complexidade ao entendimento dos mecanismos patogênicos associados.3 Além da associação de determinados genes com fenótipos específicos, estudos têm demonstrado a existência de certa correlação genótipo‐fenótipo com variantes distintas de um mesmo gene. Tais resultados apontam que a natureza e a posição da variante patogênica, bem como sua consequência em nível de mRNA e proteína, estão correlacionados às manifestações clínicas da doença.7–9 A expressividade fenotípica na EB é altamente variável, não apenas entre os diferentes subtipos, mas também dentro de cada um deles. O espectro da EB inclui pacientes com discretos sinais cutâneos, muitas vezes quase imperceptíveis, até pacientes que apresentam lesões cutâneas e extracutâneas graves, causadas por sério acometimento da adesão derme‐epiderme.10
O principal sinal clínico da EB é a formação de bolhas na pele nos locais de trauma mecânico. A depender da camada de clivagem da pele, as bolhas podem ser mais superficiais e resultar em erosões, como no caso da EBS, ou podem ser mais profundas e levar a ulcerações, como nos casos da EBJ, EBD e EBK. A distribuição das bolhas pode se dar de maneira localizada, nas extremidades ou generalizada, afeta diferentes locais do corpo. As lesões na pele podem se tornar crônicas quando o trauma mecânico é permanente ou recorrente. As membranas das mucosas oral, esofágica, traqueal, geniturinária e ocular também podem ser afetadas por erosões, úlceras e cicatrizes. Em subtipos específicos de EB, também podem ocorrer alterações em unhas e cabelos.3,6 A cicatrização progressiva pode resultar em contraturas e/ou mutilações das extremidades, microstomia e estenose esofágica, características comuns em casos mais graves de EBD e EBK. Subtipos específicos de EB associados a fenótipos graves podem apresentar envolvimento de outros órgãos e sistemas, tais como osteoporose, contraturas articulares, cardiomiopatia, amiloidose renal, atraso no crescimento, entre outros.3,6
Critérios de classificação da epidermólise bolhosa hereditáriaA classificação da EB é deveras complexa, uma vez que compreende uma gama de fenótipos com diversos níveis de gravidade associados a alterações em um número significativo de genes. Desse modo, pesquisadores especialistas em EB fazem reuniões internacionais periodicamente a fim de elaborar e atualizar uma classificação consensual para EB. A classificação mais atualizada, publicada em 2020, indicou 34 subtipos de EB (14 EBS, 9 EBJ e 11 EBD) que levam em consideração a camada da pele em que a clivagem ocorreu, as características fenotípicas (como distribuição e gravidade das bolhas e presença de características cutâneas e extracutâneas específicas), o padrão de herança genética, a expressão da proteína alterada e a alteração genética.2 Nas próximas seções, os principais tipos e subtipos de EB serão apresentados, com destaque para as características clínicas e as bases genéticas que constituem as diferentes formas da doença.9
Epidermólise bolhosa simplesA EBS caracteriza‐se por clivagem e formação de bolhas no nível epidérmico da pele e está associada a fenótipos com variável espectro de gravidade.11,12 Conta com um complexo background genético, está associada a alterações em pelo menos sete genes e, comparativamente aos demais tipos principais de EB, é a que apresenta a maior diversidade de subtipos clínicos, inclusive pelo menos 14 subtipos.1–3,6 A maior parte dos casos de EBS é herdada de maneira autossômica dominante, embora a herança autossômica recessiva seja mais comum em algumas regiões do mundo.2
Dados epidemiológicos mostram que a EBS é a que apresenta a maior incidência e prevalência em comparação aos demais tipos principais de EB, respectivamente 7,87 e 6,0 por um milhão de indivíduos, considerando uma amostra dos Estados Unidos.4 A forma localizada da doença, que inclui fenótipos mais brandos, é o subtipo mais comum de EBS (prevalência de 3,9/milhão).4 Uma vez que a EBS está frequentemente associada a fenótipos clínicos mais leves, é possível que exista um subdiagnóstico da doença e que, consequentemente, a prevalência desse tipo de EB seja ainda mais elevada.12
Subtipos clínicos da epidermólise bolhosa simplesA EBS é o tipo de EB que compreende o maior número de subgrupos clínicos. Na última classificação proposta por Has et al., os autores apresentaram 14 subtipos de EBS associados a sete genes.2 A tabela 1 traz a classificação dos subtipos de EBS, destaca o gene e a proteína alterados, bem como o padrão de herança.
Subtipos clínicos da epidermólise bolhosa simples (EBS)
| Tipo principal(padrão de herança) | Subtipos de EBS | Gene (proteína) |
|---|---|---|
| EBS(autossômica dominante) | EBS, localizada | KRT5 (queratina 5 – K5) |
| KRT14 (queratina 14 – K14) | ||
| EBS, intermediária | KRT5 (queratina 5 – K5) | |
| KRT14 (queratina 14 – K14) | ||
| EBS, grave | KRT5 (queratina 5 – K5) | |
| KRT14 (queratina 14 – K14) | ||
| EBS com pigmentação moteada | KRT5 (queratina 5 – K5) | |
| EBS, circinado migratório | KRT5 (queratina 5 – K5) | |
| EBS, intermediária | PLEC (plectina) | |
| EBS, intermediária com cardiomiopatiaa | KLHL24 (Kelch‐like 24) | |
| EBS(autossômica recessiva) | EBS, intermediária ou grave | KRT5 (queratina 5 – K5) |
| KRT14 (queratina 14 – K14) | ||
| EBS, intermediária | PLEC (plectina) | |
| EBS, localizada ou intermediária com deficiência de BP230 | DST (antígeno penfigoide bolhoso 1 – BP230b) | |
| EBS, localizada ou intermediária com deficiência de exofilina‐5 | EXPH5 (exofilina‐5c) | |
| EBS, intermediária com distrofia musculara | PLEC (plectina) | |
| EBS, grave com atresia pilóricaa | PLEC (plectina) | |
| EBS, localizada com nefropatiaa | CD151 (antígeno CD151d) |
Tabela modificada a partir da classificação de Has C et al., 2020.2
Em negrito estão os três subtipos mais prevalentes de EBS.
As formas localizada, intermediária e grave são as mais frequentes na EBS, apresentam juntas uma prevalência de 4,69/milhão, enquanto todas as outras formas de EBS atingem juntas uma prevalência de 1,31/milhão.4 Os três subtipos principais são causados por variantes patogênicas nos genes KRT5 ou KRT14, que codificam as queratinas 5 e 14, respectivamente, e representam 60%‐70% dos casos de EBS.1 A EBS localizada, anteriormente conhecida como EBS‐Weber‐Cockayne, é caracterizada pela distribuição das bolhas limitada às regiões das palmas das mãos e solas dos pés. As lesões também podem surgir em outras regiões de trauma recorrente e tendem a piorar em meses mais quentes. As bolhas geralmente não estão presentes ao nascimento, mas se desenvolvem precocemente na infância, especialmente quando a criança começa a engatinhar e caminhar. Ceratodermia palmoplantar focal (espessamento da pele das mãos e dos pés) pode aparecer em alguns casos durante a vida adulta. Entre as complicações que podem surgir, as infecções secundárias às lesões bolhosas nos pés são as mais comuns (fig. 2).3,11,12

Aspectos clínicos da epidermólise bolhosa simples (EBS). (A‐C) Ceratoderma plantar, unhas distróficas e espessas em mãos e pés de pacientes com EBS localizada; (D‐F) bolhas e erosões distribuídas de maneira generalizada em pacientes com EBS intermediária. A presença de nevos da EB é comum em todos os subtipos de EBS.
A EBS grave, antes designada como generalizada grave ou EBS‐Dowling‐Meara, apresenta um fenótipo clínico único, com bolhas grandes generalizadas que podem ocorrer de maneira agrupada (bolhas herpetiformes) e podem ser hemorrágicas.12 Durante a infância, as bolhas se desenvolvem por todo o corpo, atingem mais frequentemente as mãos e os pés, a região ao redor da boca e as regiões do tronco e pescoço. É comum nesse subtipo o envolvimento da mucosa oral, a ocorrência de ceratose palmoplantar progressiva e a distrofia de unhas.11 As lesões comumente curam sem cicatrizes, mas pode ocorrer inflamação, especialmente das bolhas hemorrágicas, que é seguida de mília e hipo‐ e hiperpigmentação da pele. Como nos demais subtipos de EBS, as bolhas tendem a melhorar com a idade; entretanto, a ceratodermia palmoplantar é mais grave para a maioria dos casos (fig. 2).3,11,12
A EBS intermediária, anteriormente conhecida como EBS generalizada intermediária ou EBS‐Koebner, frequentemente se manifesta ao nascimento com a distribuição generalizada de bolhas, mas sem a formação de agrupamento herpetiforme. A apresentação clínica é mais leve em comparação à forma grave, não envolve acometimento extracutâneo. Ainda que a distribuição seja generalizada, as bolhas atingem predominantemente as mãos e os pés. Ceratodermia palmoplantar focal pode ocorrer e as lesões tendem a piorar com o calor.3,11,12
Esses três subtipos principais de EBS são causados por alterações nos genes que codificam as queratinas 5 e 14 do citoesqueleto dos queratinócitos, como será mais detalhado na próxima seção. Além deles, outras formas de EBS também merecem destaque por representar uma fração considerável de todos os casos. Variantes patogênicas no gene PLEC, que codifica a proteína plectina, podem resultar em diferentes subtipos com distintos padrões de herança, que juntos representam até 8% dos casos de EBS.1,13 A forma autossômica dominante resulta em EBS intermediária, enquanto a forma recessiva pode resultar em EBS intermediária, EBS intermediária com distrofia muscular e EBS grave com atresia pilórica.2 A EBS intermediária com distrofia muscular é caracterizada por ocorrência de bolhas desde o nascimento e fraqueza muscular progressiva de início tardio.14,15 A EBS grave com atresia pilórica apresenta como principais características a ocorrência de bolhas no período neonatal, a presença de atresia pilórica e, frequentemente, a morte prematura.15,16 Em contraste, o subtipo com herança autossômica dominante, antes conhecido como EBS‐Ogna, agora apenas EBS intermediária, caracteriza‐se por um quadro clínico brando que inclui leve fragilidade da pele e ausência de envolvimento de outros órgãos.15,17,18
Em 2016, dois estudos independentes identificaram, quase simultaneamente, variantes patogênicas no gene KLHL24, codificante da proteína Kelch‐like 24, em 15 pacientes com fenótipo compatível com EB não relacionados entre si.19,20 A partir de então, outros pesquisadores passaram a investigar possíveis variantes nesse gene em pacientes que permaneciam até aquele momento sem a mutação genética identificada. Diversos casos com sinais típicos de EB e mutações em KLHL24 foram, então, descritos.21–25 Estima‐se que 5% dos casos de EBS sejam associados a mutações nesse gene.1 Os sinais clínicos típicos desse subtipo de EBS ainda estão sendo caracterizados, à medida que mais estudos e mais pacientes têm sido descritos. Os relatos iniciais apontavam que o fenótipo envolveria quase exclusivamente a fragilidade da pele, com lesões desde o nascimento, especialmente em pernas, e formação de bolhas em tronco e membros superiores.21 A partir de relatos adicionais, um fenótipo mais sindrômico parece estar relacionado, com envolvimento extracutâneo que incluiria uma forte associação com cardiomiopatia.23,24 Estudos demonstraram que 85% dos pacientes com variantes patogênicas em KLHL24 apresentam algum envolvimento cardíaco, evidenciado pela presença elevada de biomarcadores ou por comprovação de cardiomiopatia dilatada (40%).25,26 Problemas neurológicos, tais como atraso no desenvolvimento, deficiência intelectual, problemas de memória e miopatia, também são descritos em alguns casos.26 Futuros estudos serão indispensáveis para uma caracterização clínica completa desse novo subtipo de EBS, designado na mais recente classificação de EB como EBS intermediária com cardiomiopatia.2
Os demais subtipos de EBS descritos na tabela 1 são considerados raros, nenhum deles representa mais do que 1% do total de casos de EBS.1 Alguns apresentam sinais clínicos específicos, como pigmentação moteada e placas circinadas de carácter migratório nos subtipos que levam esses nomes. Outros subtipos se caracterizam por sinais clínicos com diferentes níveis de gravidade, que se sobrepõem a outras formas da doença.3 Para esses casos, o exame clínico não é suficiente para o diagnóstico – é necessária a aplicação de técnicas adicionais para identificação da proteína alterada ou da variante genética patogênica.27
Genética da epidermólise bolhosa simplesAinda que a expressividade clínica seja variável entre as diferentes formas de EBS, quase todas são causadas por alterações em genes que codificam componentes dos queratinócitos importantes para a organização do citoesqueleto e para as junções célula‐célula.12,28 Desse modo, a maior parte das proteínas associadas à EBS, detalhadas a seguir, têm como principal função fornecer suporte estrutural aos queratinócitos da epiderme (fig. 3).12

Representação dos complexos e proteínas associadas aos diferentes subtipos de epidermólise bolhosa simples (EBS). Alterações em componentes estruturais dos hemidesmossomos, dos filamentos intermediários de queratina e do transporte de vesículas celulares estão por trás do espectro de fenótipos da EBS. Apenas as proteínas associadas à EBS são apresentadas na figura.
As queratinas são proteínas do citoesqueleto que fornecem estabilidade mecânica às células e aos tecidos epiteliais. Os filamentos de queratina se inserem entre os desmossomos e hemidesmossomos e formam uma rede que possibilita não apenas a ligação célula‐célula dos queratinócitos, mas também entre esses e a camada subjacente da junção epiderme‐derme.11,12 As queratinas 14 (K14) e 5 (K5) são proteínas dos filamentos intermediários do tipo I e II, respectivamente, expressas nos queratinócitos basais na epiderme. No estado fisiológico, K14 e K5 formam heterodímeros de filamentos intermediários tipo I/tipo II, dos quais milhares deles se agrupam e formam o citoesqueleto de filamentos intermediários de queratina (fig. 4A).12

(A) Representação esquemática da rede de filamentos intermediários de queratina. Os filamentos de queratina se conectam aos hemidesmossomos para garantir a ligação à membrana basal subjacente e aos desmossomos a fim de assegurar o contato célula‐célula nos queratinócitos. Uma representação da configuração molecular do heterodímero K5/K14, a menor subunidade que forma os filamentos intermediários, é mostrada na parte debaixo da figura. (B) Organização dos domínios das queratinas. As queratinas apresentam um domínio central rod em α‐hélice que contém quatro segmentos (1A‐B, 2A‐B), os quais são interrompidos por três domínios de ligação (L1, L12 e L2). O domínio rod tem, em suas extremidades, motivos altamente conservados (HIP e HTP), frequentemente associados aos casos mais graves de EBS grave. Variantes nos domínios de ligação estão geralmente associadas à EBS localizada, enquanto na forma intermediária da doença as variantes envolvidas tendem a estar distribuídas pelos segmentos do domínio rod.
Variantes patogênicas nos genes KRT14 e KRT5, que codificam, respectivamente, K14 e K5, prejudicam a montagem da rede de filamentos intermediários no citoplasma dos queratinócitos, resultam no fenótipo de EBS.8 As substituições de aminoácidos (variantes missense) são o principal tipo de alteração genética nesses genes e o padrão de herança é autossômico dominante, com algumas exceções. A gravidade clínica da EBS está correlacionada, na maioria dos casos, ao local das alterações genéticas. Assim, variantes nas extremidades altamente conservadas dos domínios rod de α‐hélice, essenciais para a montagem dos filamentos de queratina, estão frequentemente associadas aos casos mais graves da doença (EBS grave) (fig. 4B).8,29 Entretanto, essa correlação não é verdadeira para todos os casos, uma vez que alguns resíduos podem ser mais ou menos importantes para a proteína, e a natureza dos aminoácidos substituídos, como sua estrutura e polaridade, também influencia no fenótipo.8,30–32 A forma mais leve, EBS localizada, está comumente associada a variantes na região de ligação (linker), enquanto a forma intermediária (EBS intermediária) compreende variantes que tendem a se distribuir ao longo das sequências de K5 e K14.8,30,33
PlectinaA plectina, codificada pelo gene PLEC, é uma proteína de ligação que desenvolve papel essencial na estabilização e organização das redes de filamentos de queratina nas células. Embora seja capaz de interagir com filamentos de actina e microtúbulos, a principal interação da plectina é com os filamentos de intermediários de queratina. Ela ancora os filamentos intermediários a sítios estratégicos, tais como as adesões focais, os desmossomos, os hemidesmossomos e outras estruturas de adesão, assim estabiliza a rede e as células.34,35 Existem diversas isoformas de PLEC, as quais são preferencialmente expressas em tecidos específicos distintos.15
Variantes patogênicas no gene PLEC podem levar a diferentes subtipos de EBS: a) EBS intermediária com padrão de herança autossômica dominante; b) EBS intermediária com padrão de herança autossômica dominante; c) EBS intermediária com distrofia muscular; e d) EBS grave com atresia pilórica, os dois últimos também com padrão de herança recessiva.2 Até o momento não está estabelecida uma clara correlação genótipo‐fenótipo em relação à posição das variantes em PLEC. A maior parte dos casos de EBS com distrofia muscular e de EBS com atresia pilórica envolve substituições que levam a códon de parada prematura (variantes nonsense) ou inserções/deleções que alteram a fase de leitura (variantes frameshift), em ambos os casos resulta na redução ou completa ausência da expressão da plectina.34 Sugere‐se que o efeito das variantes para as diferentes isoformas poderia explicar a diferença fenotípica entre esses dois subtipos (EBS com distrofia muscular e EBS com atresia pilórica).34 A única variante patogênica dominante conhecida até o momento associada à EBS intermediária (antes EBS‐Ogna) é a c.5998C>T (p. Arg.2000Trp), que leva à perturbação da plectina por efeito dominante negativo.18,34,36 Simplificadamente, variantes de efeito dominante negativo são aquelas que levam à formação de uma proteína alterada que afeta negativamente a proteína normal tipo selvagem que é coexpressa dentro da célula, reduzem sua função celular.
Outras proteínas associadas e a surpresa Kelch‐símileA maior parte dos sete genes associados à EBS tem funções estruturais para a organização das camadas da pele. O gene DST, que codifica a distonina epidérmica, também conhecida como antígeno penfigoide bolhoso (BP230 ou BPAG1e), exerce um papel na regulação da adesão dos queratinócitos e no controle dos subtipos de integrinas expressos.37 A proteína exofilina‐5, codificada por EXPH5, não é um componente integral dos filamentos intermediários, dos desmossomos ou hemidesmossomos. Ainda que seu papel não seja totalmente conhecido, sugere‐se que contribua para a regulação do tráfego da membrana celular, inclusive o controle da formação e da movimentação de vesículas ao longo das redes de actina e tubulina.37
Desde a descoberta da associação do gene KLHL24 com o fenótipo de EBS, pesquisadores buscam entender o mecanismo molecular envolvido em sua patogenicidade.20,26,38 Diferentemente da maioria das proteínas associadas à EBS, a proteína Kelch‐símile 24 não é uma proteína estrutural. Todas as variantes patogênicas identificadas nos pacientes envolvem o códon de iniciação da tradução.26 Achados preliminares sugerem que essa proteína atuaria por um novo mecanismo na EB, por meio da desregulação de sua autoubiquitinação, o que levaria à alteração nos níveis dos filamentos intermediários, especialmente da queratina 14, nos queratinócitos.20,25,38
Epidermólise bolhosa juncionalA EBJ é caracterizada por clivagem e formação de bolhas dentro da lâmina lúcida da pele. A separação epiderme‐derme nesse tipo de EB resulta, portanto, de alterações genéticas que afetam as funções de componentes essenciais da zona da membrana basal.39–42 A EBJ envolve um amplo espectro fenotípico, varia de um extremo caracterizado por letalidade precoce até outro extremo com sinais clínicos tão sutis que são quase indistinguíveis dos subtipos mais leves de EB.3 Mutações em sete genes distintos levam aos diferentes subtipos de EBJ, todos associados ao padrão de herança autossômico recessivo.2,3
A EBJ é, em comparação com os demais tipos principais de EB, a forma mais rara da doença, com uma incidência estimada em 2,68/milhão de nascidos vivos e uma prevalência de 0,5/milhão.4 Uma vez que as formas mais graves estão associadas à letalidade precoce, é provável que as estimativas de incidência e prevalência estejam subestimadas.4
Subtipos clínicos da epidermólise bolhosa juncionalA EBJ é classificada em nove subtipos, de acordo com os sinais clínicos apresentados e a gravidade do fenótipo. Os subtipos mais frequentes de EBJ são causados por variantes patogênicas nos genes que codificam a proteína laminina‐332 e o colágeno tipo XVII. O tipo de alteração genética e sua consequência para a expressão da proteína estão diretamente ligados à gravidade fenotípica, como será abordado na próxima seção. A tabela 2 apresenta os subtipos da EBJ, com destaque para os genes e as respectivas proteínas associadas.2
Subtipos clínicos da epidermólise bolhosa juncional (EBJ)
| Subtipos de EBJa | Gene (proteína) |
|---|---|
| EBJ, grave | LAMA3, LAMB3, LAMC2 (laminina‐332) |
| EBJ, intermediária | LAMA3, LAMB3, LAMC2 (laminina‐332) |
| EBJ, intermediária | COL17A1 (colágeno XVII) |
| EBJ com atresia pilóricab | ITGB4, ITGA6 (integrina α6β4) |
| EBJ, localizada | COL17A1 (colágeno XVII) |
| ITGB4 (integrina α6β4) | |
| LAMA3, LAMB3, LAMC2 (laminina‐332) | |
| ITGA3 (integrina subunidade α3) | |
| EBJ, inversa | LAMA3, LAMB3, LAMC2 (laminina‐332) |
| EBJ, início tardio | COL17A1 (colágeno XVII) |
| EBJ, síndrome LOC | LAMA3A (laminina α3A) |
| EBJ com doença pulmonar intersticial e síndrome nefróticab | ITGA3 (integrina subunidade α3) |
Tabela modificada de Has C et al., 2020.2
Subtipos em negrito são os mais frequentes grupos de EBJ.
LOC, laryngo‐onycho‐cutaneous.
A forma mais grave desse grupo é a EBJ grave, anteriormente designada como EBJ generalizada grave ou EBJ‐Herlitz, que se caracteriza por uma apresentação clínica de extensa fragilidade da pele e das membranas mucosas.2,3 As bolhas e erosões mucocutâneas estão presentes desde o nascimento e se desenvolvem por toda a superfície corporal, resultam na perda de proteínas, fluidos e ferro, o que aumenta a suscetibilidade a infecções (fig. 5).9 Tecido de granulação exuberante é formado especialmente ao redor de nariz e boca, nas nádegas e nas pregas das unhas.43 Os pacientes sofrem de dor extrema e as consequências em longo prazo incluem atraso no desenvolvimento, dificuldade de cicatrização de feridas, anemia, complicações respiratórias e infecções que levam à morte nos primeiros anos de vida.9,41,44 Esse subtipo de EBJ está associado à completa ausência ou à profunda redução da laminina‐332, como será detalhado na seção subsequente.3,9

O subtipo EBJ intermediária, antes conhecido como EBJ generalizada intermediária ou EBJ generalizada não Herlitz, compreende um grupo clinicamente heterogêneo, com distribuição generalizada de bolhas que cicatrizam e deixam sutil atrofia e hipopigmentação nos locais de lesão (fig. 5).2 A gravidade da doença é atenuada em relação ao subtipo mais grave, ainda que as membranas também possam ser afetadas.9 Outras manifestações incluem alopecia, defeitos de esmalte e distrofia ou ausência de unhas, em diferentes níveis de gravidade. Tecido de granulação, formação de feridas crônicas e envolvimento extracutâneo em córnea, laringe e trato urinário também podem estar associados.3,9,41,42 Nesse subtipo de EBJ, a expressão da laminina‐332 ou do colágeno tipo XVII está reduzida, mas não completamente ausente.9,45
A forma localizada de EBJ foi, por muito tempo, considerada uma condição rara. O aprimoramento das técnicas de análise molecular possibilitou a identificação de casos com apresentação clínica atípica em relação aos subtipos de EB mais conhecidos e, a partir de então, uma série de novos casos de EBJ localizada tem sido descrita.42,45–47 Nesse subtipo, as bolhas se distribuem de maneira localizada, principalmente nas extremidades. A EBJ localizada, assim como o subtipo intermediário, está associada à distrofia ou ausência de unhas, à hipoplasia do esmalte dentário e à tendência para o desenvolvimento de cáries (fig. 5).3,48 Contrastando com os demais subtipos, os pacientes com essa forma de EBJ usualmente não apresentam cicatrizes atróficas extensas, alopecia e outros achados extracutâneos como anemia, atraso no desenvolvimento, alterações oculares ou no trato respiratório e geniturinário.3,48 A variabilidade fenotípica é marcante na EBJ; em contraste com a forma grave da doença, a forma localizada pode apresentar um fenótipo tão leve que o único sinal clínico é a distrofia de unhas.45
Entre os subtipos mais raros de EBJ, destaca‐se a forma inversa, na qual a manifestação cutânea se dá predominantemente em áreas intertriginosas (áreas da pele que se encostam ou se atritam) e usualmente com uma extensão maior em relação à EBJ localizada.48 O subtipo EBJ com atresia pilórica envolve alterações nos genes que codificam a integrina α6β4 e está associado a fenótipos graves que envolvem, além da atresia pilórica, a distribuição generalizada de bolhas, a ausência congênita de pele e a letalidade precoce.3,49 Entretanto, a EBJ com atresia pilórica apresenta uma heterogeneidade clínica e também está associada a casos menos graves.50
Genética da epidermólise bolhosa juncionalA zona da junção epiderme‐derme compreende uma membrana basal especializada que liga a epiderme à derme e, assim, fornece à pele integridade e resistência contra forças mecânicas externas.9,51 A membrana é formada principalmente por redes de laminina‐332 e colágeno IV, que se sobrepõem e se conectam por ação de outras moléculas.9,52,53 Diversas proteínas da superfície celular dos queratinócitos e da matriz extracelular se inserem nessa rede e interagem de maneira específica entre si, garantem a adesão das camadas da pele.9,52,54
Variantes patogênicas nos genes que codificam os componentes da junção epiderme‐derme resultam na ausência ou perturbação de suas funções e na consequente diminuição da adesão das camadas da pele, o que leva aos fenótipos característicos da EBJ.9,52
Laminina‐332A laminina‐332, anteriormente conhecida como laminina‐5, é uma proteína heterotrímera constituída de cadeias α3, β3 e γ2, codificadas pelos genes LAMA3, LAMB3 e LAMC2, respectivamente. Ela é uma das proteínas essenciais da junção epiderme‐derme e é considerada uma ponte molecular entre os queratinócitos basais da epiderme e a derme subjacente.9,55 Para se conectar à superfície dos queratinócitos basais, a laminina‐332 interage com a integrina α6β4 nos hemidesmossomos e com a integrina α3β1 nas adesões focais. No lado da derme, a ligação à membrana basal é feita graças a sua interação com o colágeno tipo VII nas fibrilas de ancoragem, o que garante a adesão da membrana basal à matriz extracelular da derme (fig. 6). É por meio dessas interações proteína‐proteína que a laminina‐332, portanto, atua como um suporte para a adesão epiderme‐derme.9,41,52

Representação das principais estruturas envolvidas na adesão das camadas da pele. As principais proteínas associadas à epidermólise bolhosa juncional (indicadas em negrito, à direita da figura) interagem de modo a permitir a ligação dos filamentos intermediários de queratina às fibrilas de ancoragem (formadas por colágeno tipo VII) na derme.
Variantes patogênicas nos genes LAMA3, LAMB3 ou LAMC2 levam à ausência ou à redução da laminina‐332, o que resulta na perda de sua função como ponte entre os hemidesmossomos e as fibrilas de ancoragem. Como consequência, epiderme e derme se separam ao nível da lâmina lúcida, resultam nos fenótipos típicos da EBJ.52 A gravidade fenotípica e, por conseguinte, o subtipo clínico de EBJ estão relacionados à expressão da laminina‐332. A forma mais grave, EBJ‐grave, é usualmente causada por variantes de perda de função (variantes nulas), como as substituições que geram um códon de parada prematura (variantes nonsense) e as que alteram a fase de leitura (variantes frameshift), assim resulta na completa ausência da laminina‐332.9,41
Em cerca de 80% dos casos graves, o gene associado é o LAMB3, que codifica a cadeia β3da proteína.41 Nas formas mais leves de EBJ (intermediária e localizada), ao menos uma das variantes patogênicas associadas permite a expressão, mesmo que parcial, da proteína funcional, como as substituições de aminoácidos (missense) ou as variantes em sítios de splice que não alteram a fase de leitura (in‐frame).9,39,40
Colágeno XVIIO colágeno tipo XVII, codificado pelo gene COL17A1, é uma proteína transmembrana com orientação tipo II, na qual a extremidade N‐terminal é citoplasmática e a C‐terminal atravessa a lâmina lúcida em direção ao lado externo da célula.46 Essa proteína é um trímero de três cadeias α1, cuja principal função é conectar a rede de filamentos intermediários de queratina à membrana basal, por meio da interação com as proteínas plectina, BP230 e integrina α6β4 (fig. 6).45,46,56
Variantes patogênicas no gene COL17A1 resultam em um espectro variável de fenótipos. Ainda que diversos estudos tenham buscado possíveis correlações genótipo‐fenótipo, a única correlação clara é que os níveis de expressão do colágeno XVII influenciam diretamente a expressão clínica. Assim, pacientes com variantes genéticas que levam à completa ausência da proteína na pele, como as nonsense e as frameshift, apresentam um fenótipo grave. Enquanto isso, pacientes com sintomas mais leves da doença usualmente carregam variantes missense ou variantes em sítios de splice in‐frame, que reduzem, mas não abolem, a expressão do colágeno XVII, ou mesmo que alteram a proteína sem afetar sua expressão.45,46,57
Integrinas α6β4 e α3β1A integrina α6β4 é um polipeptídeo transmembrana heterodimérico localizado no centro dos hemidesmossomos (fig. 6).58 Sua principal função é conectar a placa hemidesmossomal dos queratinócitos basais à membrana basal subjacente e, assim, fornecer resistência mecânica à pele e às membranas mucosas.58,59 Os fenótipos resultantes de variantes patogênicas nos genes ITGA6 e ITGB4, que codificam a integrina α6β4, também são bastante heterogêneos. As formas letais da doença usualmente resultam de mutações nulas (como substituições nonsense e inserções e deleções frameshift) em ambos os alelos, enquanto as formas mais leves usualmente envolvem a presença de substituições de aminoácidos (variantes missense) em ao menos um dos alelos.50
O gene que codifica a subunidade α3da integrina α3β1 foi recentemente associado a uma forma rara de EBJ (EBJ com envolvimento respiratório e renal).60 Ainda que seja conhecida a importância da integrina α3β1 para o epitélio, inclusive os queratinócitos epidérmicos, as células epiteliais alveolares e os podócitos, os mecanismos moleculares envolvidos nesse subtipo de EBJ ainda não são conhecidos.61
Epidermólise bolhosa distróficaA EBD é caracterizada pela clivagem da pele na região da sublâmina densa, na porção superior da derme.3 A EBD pode ser herdada tanto com padrão autossômico dominante quanto autossômico recessivo. Ela está associada a um amplo espectro fenotípico e de gravidade, que varia desde a ocorrência isolada de leve distrofia de unhas até a formação generalizada de bolhas com cicatrização mutilante de mãos e pés, envolvimento extracutâneo grave e morte prematura.3,7,62 Ainda que a maior parte das variantes patogênicas da EBD seja típica do padrão de herança dominante ou do recessivo, algumas alterações genéticas já foram descritas em ambas as formas, o que adicionou complexidade ao entendimento das bases moleculares desse tipo de EB.3,63 O gene COL7A1, que codifica o principal constituinte das fibrilas de ancoragem, o colágeno tipo VII, é o único gene conhecido associado à EBD.
A EBD é classificada em dois grupos principais, de acordo com o padrão de herança associado – EBD dominante (EBDD) e EBD recessiva (EBDR) –, e subdividida em pelo menos 11 subtipos clínicos, que levam ainda em consideração as características fenotípicas envolvidas.2 A incidência e prevalência da EBD foram estimadas em 6,65 e 3,3/milhão de nascidos vivos, respectivamente. A prevalência das formas dominante e recessiva é bastante similar, 1,5 e 1,4/ milhão, nessa ordem; os casos de EBD sem padrão de herança definido atingem uma prevalência de 0,4/milhão.4
Subtipos clínicos da epidermólise bolhosa distróficaA classificação da EBD em seus diversos subtipos leva em consideração os sinais cutâneos e extracutâneos característicos, a distribuição das bolhas (localizada ou generalizada) e a gravidade do fenótipo.2,3 Ainda que alguns subtipos se caracterizem por sinais clínicos específicos e de gravidade distinta, existe uma sobreposição de certos fenótipos entre alguns grupos (fig. 7 A‐I), o que dificulta o diagnóstico e exige testes moleculares adicionais.3,27 Levando em consideração que a EBD é herdada tanto de maneira dominante quanto recessiva, a classificação do tipo de EBD é especialmente importante para fins de aconselhamento genético.6 A tabela 3 apresenta os subtipos de EBD.

Aspectos clínicos da epidermólise bolhosa distrófica (EBD). (A) Paciente com EBD dominante pré‐tibial. (B‐C) Distrofia das unhas de pés e mãos de pacientes com EBD dominante localizada. (D‐E) Pseudosindactilia (fusão dos dedos) e distribuição generalizada de bolhas e extensas cicatrizes em paciente com EBD recessiva grave. (G‐H) Bolhas e cicatrizes generalizadas especialmente em joelhos, pernas, pés e mãos em paciente com EBD recessiva intermediária; a hiperqueratose das mãos e dos pés leva a contraturas de flexão. (F, I) Bolhas e lesões restritas a mãos e pés e distrofia de unhas em pacientes com EBD recessiva localizada.
Subtipos clínicos da epidermólise bolhosa distrófica (EBD)
| Tipo principal | Subtipos de EBD | Gene (proteína) |
|---|---|---|
| EBD dominante (EBDD) | EBDD, intermediária | COL7A1 (colágeno tipo VII) |
| EBDD, localizadaa | COL7A1 (colágeno tipo VII) | |
| EBDD, pruriginosa | COL7A1 (colágeno tipo VII) | |
| EBDD, autorreparávelb | COL7A1 (colágeno tipo VII) | |
| EBD recessiva (EBDR) | EBDR, grave | COL7A1 (colágeno tipo VII) |
| EBDR, intermediária | COL7A1 (colágeno tipo VII) | |
| EBDR, inversa | COL7A1 (colágeno tipo VII) | |
| EBDR, localizadac | COL7A1 (colágeno tipo VII) | |
| EBDR, pruriginosa | COL7A1 (colágeno tipo VII) | |
| EBDR, autorreparávelb | COL7A1 (colágeno tipo VII) | |
| EBD dominante e recessiva (heterozigose composta) | EBD, grave | COL7A1 (colágeno tipo VII) |
Tabela modificada de Has C et al., 2020.2
Em negrito estão os subtipos mais frequentes de EBD.
Nos subtipos de EBD, a gravidade dos fenótipos está relacionada, em certo nível, à consequência das mutações de COL7A1 para a formação das fibrilas de ancoragem, as quais podem apresentar alterações morfológicas, ter sua expressão reduzida ou mesmo estar completamente ausentes, como será detalhado na próxima seção.64 Ainda que as formas graves de EBD sejam descritas como as mais frequentes, é possível que haja um subdiagnóstico das formas localizadas, dado o envolvimento de sinais clínicos mais sutis. A partir do desenvolvimento das tecnologias de sequenciamento de nova geração, novos estudos, inclusive grandes números amostrais, têm permitido estabelecer correlações genótipo‐fenótipo para a doença.34–67 Os principais subtipos de EBD são detalhados a seguir.
A EBDR grave, antes denominada EBDR generalizada grave ou EBDR‐Hallopeau‐Siemens, constitui a forma mais grave de EBD, usualmente associada à ausência completa da expressão do colágeno VII.68 Os sinais clínicos clássicos envolvem a formação de bolhas generalizadas pelo corpo desde o nascimento e cicatrizes mutilantes que levam à pseudosindactilia (fusão dos dedos) em mãos e pés e que resultam em contraturas das articulações. Além disso, úlceras crônicas de difícil cicatrização, mília e distrofia de unhas são outros sinais típicos (fig. 7 D‐E). As bolhas podem ocorrer na mucosa oral, na região da córnea e no epitélio gastrintestinal; estenose esofágica é descrita em muitos casos. Anemia, atraso no desenvolvimento e carcinoma de células escamosas associado à morte precoce ainda compõem o padrão fenotípico desse subtipo de EBDR.3,62,64
A EBDR intermediária, anteriormente conhecida como EBDR generalizada intermediária ou EBDR não Hallopeau‐Siemens, também se caracteriza pela formação de bolhas de distribuição generalizada, entretanto envolve menor gravidade (fig. 7 G‐H).69 Sinais clínicos muito frequentes na forma grave, como a constrição esofágica, as lesões na córnea e a pseudosindactilia de mãos e pés, ainda ocorrem, mas são menos usuais na forma intermediária da doença. Atraso no desenvolvimento e anemia são condições ainda mais raras nesse subtipo de EBDR.3,69
Entre os subtipos dominantes, a forma intermediária, EBDD intermediária (antes EBDD generalizada), caracteriza‐se pela formação de bolhas com distribuição generalizada ao nascimento, lesões albopapuloides, mília, cicatrização atrófica e distrofia de unhas. Em contraste com a forma EBDR grave, atraso no desenvolvimento, anemia grave e risco de carcinoma de células escamosas não são características dos subtipos de EBDD.3,69
Em meio à complexidade que é a classificação da EBD em seus diversos subtipos, as formas localizadas de ambos os padrões de herança dificultam ainda mais: a EBDD localizada e a EBDR localizada são clinicamente indistinguíveis.3 Ou seja, para fenótipos típicos de EB distrófica localizada, o resultado genético é imprescindível para a definição do padrão de herança associado e, em última análise, para o correto aconselhamento genético. Trata‐se de subtipos com fenótipos muito atenuados de EB, cujo principal sinal clínico envolve a distribuição de bolhas de maneira localizada nas regiões de mãos e pés e distrofia de unhas (fig. 7 B, C, F e I). Mília e cicatrizes atróficas também podem ocorrer. Não há envolvimento extracutâneo e nem pseudosindactilia nessas formas da doença.3,69
A subdivisão da EBD ainda abrange uma série de outros subtipos mais raros, que geralmente envolvem sinais clínicos específicos. A EBDR inversa caracteriza‐se por uma sucessão peculiar de achados clínicos que se inicia com a formação de bolhas generalizadas no período neonatal que, no início da infância, se curam e deixam cicatrizes atróficas. Os sinais cutâneos tendem a melhorar com o passar dos anos, enquanto lesões graves se desenvolvem nas mucosas oral, esofágica, anal e genital.27,70 A EBD pruriginosa compreende um subtipo generalizado que inclui a queixa de prurido grave.69,71 A EBD autorreparável, antes conhecida como EBD dermólise bolhosa do recém‐nascido, é um subtipo de EBD que se apresenta ao nascimento, ou logo após, com a formação de bolhas generalizadas. Contrastando com os demais subtipos, os sinais clínicos dessa forma da doença deixam de aparecer dentro de 6 a 24 meses de vida.69
Genética da epidermólise bolhosa distróficaA forma distrófica da EB se caracteriza pela separação do tecido logo abaixo da lâmina densa, no nível das fibrilas de ancoragem. Como será detalhado a seguir, o principal constituinte das fibrilas de ancoragem é o colágeno VII, codificado a partir do gene COL7A1. Alterações nesse gene resultam em modificações morfológicas ou redução/ausência das fibrilas de ancoragem, levam aos diferentes fenótipos de EBD.72 O nível de expressão do COL7A1 nos pacientes com EBD está inversamente correlacionado com a gravidade fenotípica.7,73
Colágeno tipo VIIO colágeno tipo VII é o principal, se não o único, componente das fibrilas de ancoragem. Essas, por sua vez, são grandes estruturas em forma de U que se estendem da parte de baixo da membrana basal derme‐epiderme até a camada superior da derme (derme papilar).52 Desse modo, as fibrilas de ancoragem fornecem integridade para a associação da epiderme à derme subjacente.62
O COL7A1 é um gene que apresenta um arranjo complexo, consiste em 118 éxons e um tamanho de aproximadamente 32kb. Esse gene codifica polipeptídeos de colágeno pro‐α1 (VII), que irão se organizar em fibrilas de ancoragem.62,74 A estrutura desses polipeptídeos inclui um domínio colagenoso em sua porção central, formado por repetições Gly‐X‐Y que se dobram para atingir a conformação de tripla hélice característica das moléculas de colágeno.62 O domínio de tripla hélice do colágeno VII contém 19 interrupções ou imperfeições, inclusive um segmento central não colagenoso de 39 aminoácidos.74 Tais interrupções na sequência Gly‐X‐Y fornecem flexibilidade às moléculas de colágeno VII.52 O domínio central colagenoso é flanqueado por domínios globulares não helicoidais, o domínio NC1 na extremidade amino‐terminal e o domínio NC2 na extremidade carboxi‐terminal.52,62,74
A figura 8 apresenta um esquema de formação das fibrilas de ancoragem. Os polipeptídeos de colágeno pro‐α1 (VII) são sintetizados principalmente pelos queratinócitos da epiderme, mas também pelos fibroblastos da derme.52 Três polipeptídeos formados se associam por meio de suas extremidades carboxi‐terminais (domínios NC2), enquanto seus domínios colagenosos se enovelam em uma conformação de tripla hélice. Duas moléculas de colágeno tipo VII em tripla hélice formam dímeros antiparalelos e, após a remoção de parte da extremidade carboxi‐terminal (NC2), o dímero formado é estabilizado por ligações dissulfeto intermoleculares.
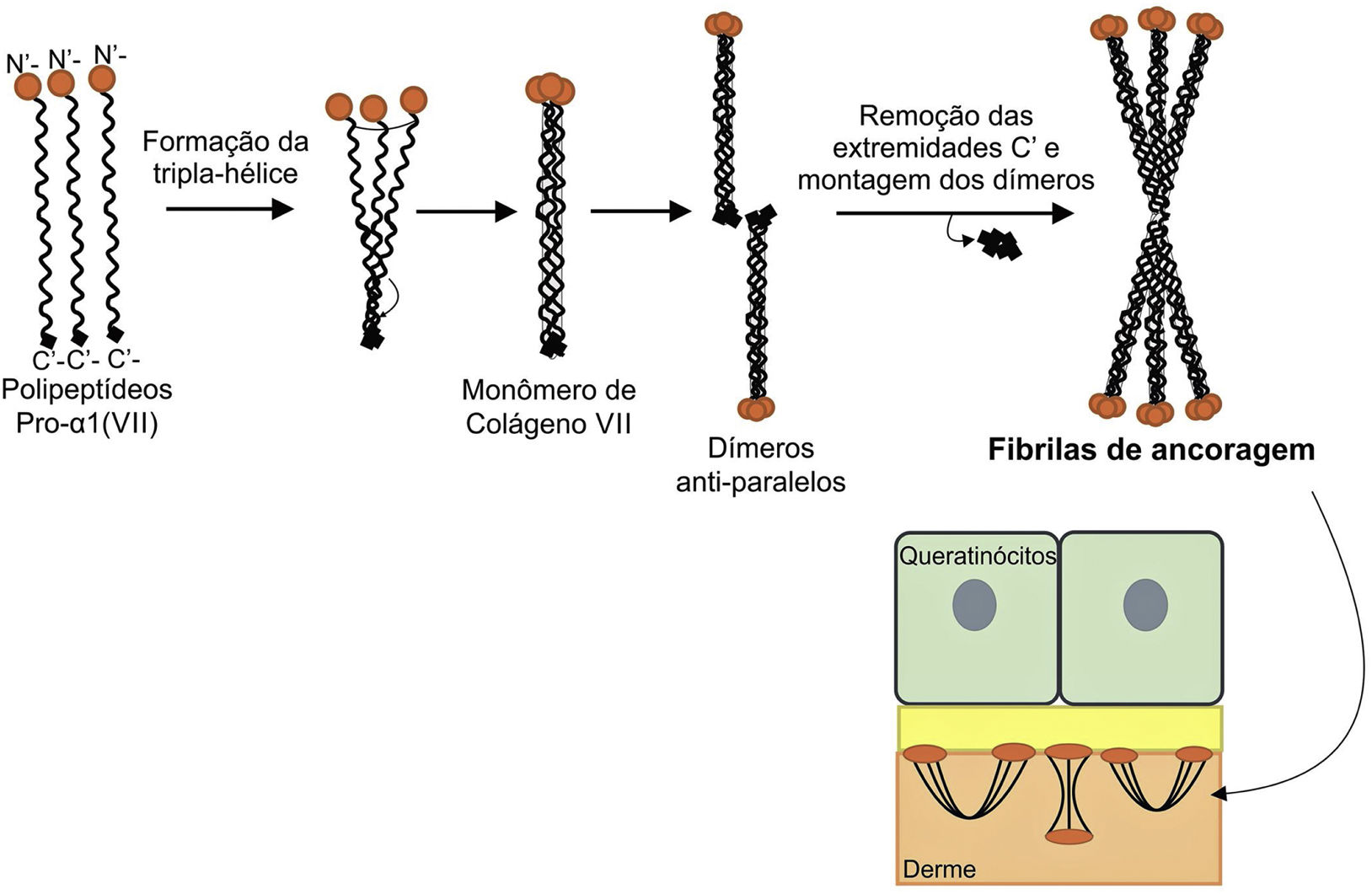
Representação esquemática da montagem das fibrilas de ancoragem a partir dos polipeptídeos de colágeno pro‐a1 (VII). Três polipeptídeos se associam pelas suas extremidades C‐terminais, enquanto seus domínios colagenosos se dobram em formação de tripla hélice. A partir de então, os monômeros de colágeno tipo VII em tripla hélice formam dímeros antiparalelos, que se unem após a remoção de parte da extremidade C‐terminal. As diversas moléculas de dímeros se juntam lateralmente e formam fibrilas de ancoragem. Os domínios N‐terminais das fibrilas de ancoragem se ligam por homologia às macromoléculas na lâmina densa, estabilizam a adesão dessa com a derme subjacente.
Subsequentemente, diversos dímeros de colágeno tipo VII se organizam lateralmente nas fibrilas de ancoragem. A ligação das fibrilas à lâmina densa ocorre graças ao domínio aminoterminal não colagenoso NC1 que, por homologia, liga‐se às proteínas da lâmina densa, estabiliza a associação dessa camada com a derme subjacente (fig. 8).62,72,75–77
A importância de se conhecer as etapas da formação das fibrilas de ancoragem reside no fato de que perturbações em quaisquer dessas etapas resultam em consequências específicas para a estrutura e expressão das fibrilas, o que leva, em última análise, aos diferentes fenótipos da EBD.62,72 Como será apresentado a seguir, pode‐se dizer que o fenótipo da EBD é, via de regra, determinado pela expressão e função residual de COL7A1, ainda que exceções a esse padrão já tenham sido relatadas.78,79
A EBDR grave é frequentemente causada por variantes de códon de parada prematura (variantes nulas) em ambos os alelos do gene COL7A1, resulta de substituições nonsense, de inserções ou deleções frameshift ou, ainda, de variantes em sítios de splice que resultam em alteração da fase de leitura.7,80 Tais alterações causam graves consequências para a proteína formada, resultam em decaimento de mRNA mediado por mutações nonsense (mecanismo de degradação de transcritos com códon de parada prematura) ou na formação de polipeptídeos truncados que não são capazes de se associar para a montagem das fibrilas de ancoragem.7,62,80
A forma EBDR intermediária, por sua vez, é frequentemente causada por variantes em heterozigose composta: uma variante de códon de parada prematura e uma substituição de aminoácidos.7 Cabe ressaltar, entretanto, que diversos outros tipos de variantes, em diferentes combinações, também já foram associadas a esse subtipo.67,79,81–84 O que caracteriza a forma intermediária é a presença de pelo menos uma variante que permite alguma produção de colágeno tipo VII, assim possibilita a montagem, ainda que parcial, das fibrilas de ancoragem. Desse modo, as alterações genéticas associadas a esse subtipo podem afetar a associação dos polipeptídeos, a formação e a estabilidade da tripla hélice ou, ainda, causar alguma modificação conformacional na proteína.7,62 Os polipeptídeos formados nesses casos ainda são capazes de formar um pequeno número de fibrilas de ancoragem, mesmo que instáveis, o que explica o fenótipo mais atenuado em relação ao subtipo grave da doença.7,85
Nesse mesmo sentido, a forma localizada da EBDR está correlacionada a variantes que afetam minimamente a formação das fibrilas de ancoragem, seja reduzindo sua expressão ou por meio de leve alteração conformacional ou de estabilidade.86–88 Curiosamente, as variantes mais descritas em casos de EBDR localizada são as que ocorrem em sítios de splice.83,86,88,89 Variantes de splice estão associadas a uma diversidade de fenótipos, depende de sua influência para a sequência da proteína formada. Na forma localizada, elas usualmente resultam na excisão de éxons inteiros (exon skipping), entretanto sem alterar o restante da sequência da proteína. Estudos têm demonstrado que essas variantes não alteram a totalidade dos transcritos formados, mas apenas uma parcela deles, possibilitam a formação de transcritos normais em quantidade considerável. Isso possibilita a montagem das fibrilas de ancoragem com apenas pequenos defeitos funcionais, o que justifica o fenótipo tão leve dos pacientes.86,88–90
A forma dominante de EBD usualmente envolve substituições de glicina no domínio colagenoso de tripla hélice, embora outras substituições, inserções, deleções e variantes em sítio de splice também tenham sido descritas.7,62,91,92 Essas variantes afetam aminoácidos essenciais para a estrutura da tripla hélice e, por isso, estão associadas à diminuição da estabilidade das fibrilas de ancoragem.7,62,72 Alguns aminoácidos estão posicionados em segmentos mais importantes para a estrutura do colágeno VII e algumas substituições afetam a formação da molécula de maneira mais significativa do que outras. Assim, a natureza e a posição da variação genética estão intimamente correlacionadas ao fenótipo resultante.7,93
Epidermólise bolhosa de KindlerA EBK, antes conhecida como síndrome de Kindler, foi apenas recentemente classificada como um dos tipos principais de epidermólise bolhosa, a partir da constatação de que as bases clínicas e biológicas envolvidas são compatíveis com EB.2,3,27 Trata‐se de um subtipo raro de EB, relatado em aproximadamente 250 indivíduos em todo o mundo, desde sua primeira descrição.2 A apresentação clínica da EBK pode mimetizar diversos subtipos de EB, o que dificulta o diagnóstico em muitos casos.27,94,95 A EBK foi diferenciada dos demais três tipos de EB por apresentar características peculiares. Em primeiro lugar, na EBK a clivagem da pele pode ocorrer em múltiplas camadas (intraepidérmica, juncional ou sublâmina densa), o que impede sua inclusão em um dos outros grupos da doença. Além disso, características clínicas distintas são muito comuns na EBK, especialmente a poiquilodermia, que combina a hiper ou hipopigmentação da pele e a ocorrência de aglomerados de vasos sanguíneos logo abaixo da epiderme (telangiectasias) e fotossensibilidade, que se manifesta como eritema e queimadura na pele após exposição ao sol.3,27,96 O fenótipo característico da EBK é usualmente progressivo e inclui, além da poiquilodermia e da fotossensibilidade, a formação de bolhas e a extensa atrofia da pele. Achados extracutâneos da EBK incluem erosões na gengiva e envolvimento ocular, esofágico, gastrintestinal e geniturinário.3,97
A EBK apresenta herança autossômica recessiva e é causada por alterações no gene FERMT1, que codifica a kindlina‐1, uma proteína associada às integrinas e às adesões focais.98,99 Na pele, a kindlina‐1 está localizada junto aos queratinócitos basais e atua como uma proteína adaptadora das adesões focais, liga os filamentos de actina às proteínas da membrana.97,100 Em pacientes com EBK, os queratinócitos se tornam desorganizados, perdem sua estrutura característica e sua polarização, além de ter sua proliferação reduzida. Estudos in vitro demonstraram que a perda da função da kindlina‐1 está associada à anormalidade no formato celular e à alteração nos processos de adesão, proliferação e motilidade das células.97,101 Essas alterações resultam de perturbações nas adesões focais que, em situações normais, garantem a ancoragem do citoesqueleto às plataformas de sinalização das integrinas.102,103 A kindlina‐1 compõe o complexo de adesão da integrina β1 e participa da formação das adesões focais, o que explica as consequências da perda de sua função para a estrutura e adesão das células da pele e justifica o fenótipo de fragilidade cutânea associado.94,97,99
Já foram descritas 84 variantes patogênicas em FERMT1 (HGMD database − www.hgmd.cf.ac.uk), a maioria delas associadas à formação de códon de parada prematura e consequente perda de função da kindlina‐1. Outras variantes, entretanto, também foram identificadas, inclusive substituições de aminoácidos, variantes em sítios de splice e variantes intrônicas.94,95,97,99 Uma vez que a maior parte das variantes é nula, resulta na ausência da kindlina‐1, correlações genótipo‐fenótipo não são bem estabelecidas para a EBK, visto que a natureza ou a posição da variante não parece influenciar significativamente a apresentação clínica. Embora os sinais clínicos da EBK sejam comuns aos pacientes com variantes nulas, existe uma evidente variabilidade clínica, o que sugere uma influência de fatores ambientais e/ou epigenéticos.97
Outras desordens de fragilidade da peleA fragilidade da pele, além de ser característica dos subtipos clássicos de EB, também está presente em outros grupos de doenças hereditárias, tais como as desordens de descamação da pele, as erosivas, as hiperceratósicas e as do tecido conectivo, apresentadas na tabela 4.2 Essas doenças se assemelham à EB no que diz respeitos às alterações nas camadas da pele e/ou nos mecanismos patogênicos envolvidos e, portanto, devem ser consideradas para o diagnóstico diferencial, especialmente em casos de recém‐nascidos.2
Outras desordens de fragilidade da pele
| Nível de clivagem na pele | Denominação | Padrão de herança | Gene (proteína) alterados |
|---|---|---|---|
| Intraepidérmico | Desordens de descamação da pele | AR | TGM5 (Transglutaminase 5)CSTA (Cistatina A)CTSB (Catepsina B)SERPINB8 (Serpina B8)FLG2 (Filagrina 2)CDSN (Corneodesmosina)CAST (Calpastatina)DSG1 (Desmogleína 1)SPINK5 (LEKTI) |
| Intraepidérmico | Desordens erosivas com fragilidade da pele | AR | DSP (Desmoplaquina)JUP (Placoglobina)PKP1 (Placofilina 1)DSC3 (Desmocolina 3)DSG3 (Desmogleína 3) |
| Intraepidérmico | Desordens hiperceratóticas com fragilidade da pele | ||
| Ictioses queratinopáticas | AD | KRT1 (Queratina 1) | |
| KRT10 (Queratina 10) | |||
| KRT2 (Queratina 2) | |||
| AR | KRT10 (Queratina 10) | ||
| Paquioníquia congênita | AD | KRT6A (Queratina 6A) | |
| KRT6B (Queratina 6B) | |||
| KRT6C (Queratina 6C) | |||
| KRT16 (Queratina 16) | |||
| KRT17 (Queratina 17) | |||
| Dérmico | Desordem sindrômica do tecido conectivo com fragilidade da pele | AR | PLOD3 (Lisil hidroxilase 3) |
Tabela modificada de Has C et al., 2020.2
AD, autossômica dominante; AR, autossômica recessiva.
Embora não estejam incluídos como EB clássica, essas desordens apresentam como marcantes características fenotípicas a fragilidade da pele e, frequentemente, das mucosas, envolvem os mesmos sinais clínicos e necessidades de saúde. Desse modo, para fins de prestação de assistência médica e socioeconômica, recomenda‐se considerar tais desordens juntamente com as formas clássicas de EB.2
Diagnóstico da epidermólise bolhosa hereditáriaComo já apresentado, a EB compreende quatro formas principais e múltiplos subtipos clínicos, que se caracterizam por uma ampla variedade de sinais clínicos. Periodicamente, pesquisadores de todo o mundo se reúnem para atualizar os critérios de diagnóstico e a classificação dos subtipos de EB.2,3,27,104,105 O objetivo da classificação é conseguir particularizar em diferentes grupos toda a heterogeneidade genética e clínica da doença, a fim de auxiliar o preciso diagnóstico e subclassificação. Entretanto, muitos dos sinais clínicos se sobrepõem entre os subtipos. Isso evidencia que a classificação da EB, em grande parte dos casos, não é tão óbvia.3,79 Na realidade, o que se nota dentro das diferentes formas de EB é um espectro contínuo de gravidade fenotípica. Casos mais graves são, em geral, mais fáceis de se classificar, enquanto os casos que se enquadram ao longo desse contínuo exigem análises adicionais, além da observação clínica apenas.
Levando em consideração os inúmeros complexos proteicos que estreitamente interagem a fim de fornecer integridade e adesão às camadas da pele, é de se esperar que diferentes alterações genéticas afetem de maneira distinta cada um desses componentes, o que acarreta consequências diversas para a estruturação da pele e, em última análise, resulta nos diversos fenótipos conhecidos da EB.5,6,106 O preciso diagnóstico e a subclassificação correta da EB possibilitam a indicação prognóstica da gravidade da doença, o aconselhamento genético para o paciente e seus parentes, o diagnóstico genético pré‐natal ou pré‐implantacional, a inclusão em ensaios clínicos e a medicina de precisão.6 A confirmação do diagnóstico de EB pode ser feita por meio de: a) mapeamento por imunofluorescência, a partir da análise de biópsia de pele; b) microscopia eletrônica de transmissão, por meio da análise da ultraestrutura da pele; ou c) análise genética direta dos genes associados à EB.1,6
O mapeamento por imunofluorescência (MIF), ou imunomapeamento, é um dos métodos empregados para a determinação do tipo principal de EB e, muitas vezes, da provável proteína que está alterada.27 O método se baseia no uso de anticorpos monoclonais contra proteínas da junção derme‐epiderme, o que possibilita que se identifique a camada da pele onde a clivagem ocorre e a abundância relativa das proteínas.107 Trata‐se de uma ferramenta importante para o diagnóstico, especialmente dos casos de maior gravidade da doença, quando a expressão da proteína alterada é ausente ou muito reduzida.107,108 Para os casos de menor gravidade, a clivagem da pele pode não ser identificada na amostra e, do mesmo modo, podem não ser observadas alterações na imunorreatividade dos marcadores usados, o que leva a frequentes resultados inconclusivos.107
A microscopia eletrônica de transmissão (MET) possibilita a detecção e a avaliação semiquantitativa de estruturas presentes nas camadas da pele, como os filamentos de queratina, os desmossomos, os hemidesmossomos, os filamentos de ancoragem e as fibrilas de ancoragem. Assim, a MET proporciona a identificação de alterações dessas estruturas em relação a sua quantidade e aparência, que se correlacionam aos distintos subtipos de EB.27,108 A interpretação dos resultados, entretanto, requer um significativo conhecimento acerca das estruturas de adesão célula‐célula e célula‐matriz e sua aparência na pele normal e na pele com EB.6 Além disso, existem poucos laboratórios no mundo que fazem a MET para análise de EB; por isso, é uma técnica pouco empregada para o diagnóstico da doença.27
O teste genético fornece o diagnóstico definitivo do subtipo de EB, o padrão de herança associado e a variante patogênica causal.27 Três métodos principais são usados para a análise genética: a) sequenciamento de Sanger; b) sequenciamento de um painel de genes por meio de sequenciamento de nova geração (do inglês, next‐generation sequencing, NGS); c) sequenciamento do exoma inteiro por NGS.6 O sequenciamento de Sanger é uma excelente abordagem quando se conhece o gene candidato, que pode ser inferido a partir de exames prévios (como MIF e MET), ou que pode ser sugerido quando o fenótipo é claramente típico de um subtipo específico. Quando não se tem a definição do gene candidato, o sequenciamento de Sanger se torna um método muito trabalhoso e de alto custo, já que envolve a análise de todos os éxons dos genes em análise.1,6 O painel de genes por NGS possibilita a análise rápida de múltiplos genes simultaneamente e é uma efetiva abordagem quando não se conhece o gene candidato.1,6 O sequenciamento do exoma completo torna possível a análise da maior parte das regiões codificadoras de proteínas e também é uma ferramenta útil quando não se conhece o gene candidato. A vantagem desse método em relação ao painel de genes é a possibilidade de descoberta de novos genes associados à doença. A desvantagem está ligada à maior complexidade para análise e interpretação dos dados, exige expertise e recursos computacionais adicionais, o que consome mais tempo até o diagnóstico final.6
Considerações finaisA EB é uma doença heterogênea que envolve amplo espectro de gravidade, distintos padrões de herança e muitos genes causais. As formas mais graves da doença apresentam sinais clínicos debilitantes, tais como mutilação de membros, inflamações e infecções recorrentes, dor crônica e letalidade precoce. Embora ainda não exista cura para a EB e os tratamentos sejam apenas sintomáticos, estudos têm demonstrado avanços para potenciais abordagens terapêuticas futuras.
Estudos clínicos e genéticos têm possibilitado compreender muitos dos mecanismos moleculares responsáveis pela adesão das camadas da pele e que estão associados aos distintos subtipos de EB. Ao menos 16 genes já são conhecidos e a classificação mais recente da doença descreveu 34 subtipos clínicos. Ainda assim, algumas lacunas permanecem a ser preenchidas acerca das bases clínicas e genéticas da EB e dos mecanismos de modulação dos fenótipos de fragilidade da pele. Estudos futuros serão importantes para o melhor entendimento da EB e para o desenvolvimento de potenciais tratamentos.
Suporte financeiroNenhum.
Contribuição dos autoresLuiza Monteavaro Mariath: Elaboração e redação do manuscrito; revisão crítica da literatura.
Juliana Tosetto Santin: Aprovação da versão final do manuscrito; revisão crítica da literatura.
Lavínia Schuler‐Faccini: Aprovação da versão final do manuscrito; revisão crítica do manuscrito.
Ana Elisa Kiszewski: Aprovação da versão final do manuscrito; revisão crítica do manuscrito.
Conflitos de interesseNenhum.
Questões EMC- 1.
A EB é classificada em quatro tipos principais, que se relacionam à camada da pele na qual ocorre a formação de bolhas. Assinale a opção que correlaciona corretamente o tipo de EB e a respectiva camada da pele em que se dá a clivagem.
- a)
EBS – intraepidérmica; EBJ – lâmina lúcida da membrana basal; EBD – padrão misto de clivagem; EBK – camada superior da derme.
- b)
EBS – camada superior da derme; EBJ – lâmina lúcida da membrana basal; EBD – intraepidérmica; EBK – padrão misto de clivagem.
- c)
EBS – lâmina lúcida da membrana basal; EBJ – intraepidérmica; EBD – camada superior da derme; EBK – padrão misto de clivagem.
- d)
EBS – intraepidérmica; EBJ – lâmina lúcida da membrana basal; EBD – camada superior da derme; EBK – padrão misto de clivagem.
- 2.
Clivagem no nível epidérmico da pele, maior diversidade de subtipos clínicos, padrão de herança predominantemente autossômico dominante, maior incidência e prevalência entre os tipos de EB. As características mencionadas se referem a qual tipo principal de EB?
- a)
EBS.
- b)
EBJ.
- c)
EBD.
- d)
EBK.
- 3.
São proteínas do citoesqueleto que fornecem estabilidade mecânica às células e tecidos epiteliais. Elas se associam em um heterodímero, formam os filamentos intermediários que tornam possível a ligação célula‐célula dos queratinócitos e a ligação dos queratinócitos à camada subjacente. O texto refere‐se a quais proteínas?
- a)
Plectina e BP230.
- b)
Colágenos VII e XVII.
- c)
Integrinas α6β4 e α3β1.
- d)
Queratinas 5 e 14.
- 4.
Qual das opções a seguir apresenta as proteínas que atuam na junção epiderme‐derme associadas à EBJ?
- a)
Colágeno VII.
- b)
Queratina 5, queratina 14, plectina, Kelch‐like 24, exofilina‐5.
- c)
Laminina‐332, colágeno XVII, integrina α6β4, integrina α3β1.
- d)
Kindlina‐1.
- 5.
Caracterizada pela clivagem da pele na região da sublâmina densa, na porção superior da derme, essa forma de EB apresenta tanto padrão de herança autossômica dominante quanto recessiva; está associada a um amplo espectro fenotípico e de gravidade, que varia desde a ocorrência isolada de leve distrofia de unhas até a formação generalizada de bolhas com cicatrização mutilante de mãos e pés, envolvimento extracutâneo grave e morte prematura. A descrição se refere a qual tipo principal de EB?
- a)
EBS.
- b)
EBJ.
- c)
EBD.
- d)
EBK.
- 6.
É a forma mais grave de EBD, cujos principais sinais clínicos envolvem a formação de bolhas generalizadas pelo corpo, pseudossindactilia, úlceras crônicas de difícil cicatrização, anemia, atraso no desenvolvimento, acometimento de mucosas e maior suscetibilidade ao desenvolvimento de carcinoma de células escamosas. A afirmação se refere a que subtipo de EB?
- a)
EBD recessiva localizada.
- b)
EBD recessiva grave.
- c)
EBD dominante intermediária.
- d)
EBJ grave.
- 7.
A EBD é causada por alterações em um único gene, o que resulta em modificações morfológicas ou redução/ausência de um importante constituinte das camadas da pele, que fornece integridade para a associação da epiderme à derme subjacente. Assinale a opção que indica a proteína associada à EBD e a estrutura de adesão por ela formada.
- a)
Colágeno XVII, fibrilas de ancoragem.
- b)
Colágeno VII, filamentos intermediários.
- c)
Colágeno VII, fibrilas de ancoragem.
- d)
Colágeno XVII, fibras de colágeno.
- 8.
Sabe‐se que o tipo de alteração genética e sua consequência para a expressão da proteína estão diretamente ligados à gravidade fenotípica da EB distrófica recessiva (EBDR). Assinale a opção incorreta.
- a)
A presença de variantes de códon de parada prematura (variantes nulas) em ambos os alelos do gene COL7A1 está frequentemente associada à ausência completa da expressão do colágeno VII, resulta na EBDR grave.
- b)
A EBDR localizada está usualmente associada a variantes que afetam minimamente a formação das fibrilas de ancoragem, seja reduzindo sua expressão ou por meio de leve alteração conformacional ou de estabilidade, o que explica o fenótipo atenuado desse subtipo de EB.
- c)
Na forma EBDR intermediária, usualmente pelo menos um dos alelos carrega uma variante que permite alguma produção de colágeno tipo VII, assim possibilita a montagem, ainda que parcial, das fibrilas de ancoragem.
- d)
A EBDR grave é principalmente causada por substituições de glicina que são essenciais para a estrutura da tripla hélice do colágeno VII e, por isso, estão associadas à diminuição da estabilidade das fibrilas de ancoragem.
- 9.
Trata‐se de um subtipo raro de EB, causado por alterações no gene FERMT1 e que envolve sinais clínicos típicos como poiquilodermia e fotossensibilidade. A afirmação refere‐se a que tipo principal de EB?
- a)
EBS.
- b)
EBJ.
- c)
EBD.
- d)
EBK.
- 10.
Qual dos métodos de análise a seguir não é usado para o diagnóstico de epidermólise bolhosa?
- a)
Cariótipo.
- b)
Microscopia eletrônica de transmissão.
- c)
Mapeamento por imunofluorescência.
- d)
Sequenciamento de nova geração.
GABARITO
Infecções cutâneas bacterianas graves. An Bras Dermatol. 2020;95(4):407–17.
Como citar este artigo: Mariath LM, Santin JT, Schuler-Faccini L, Kiszewski AE. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol. 2020;95:551–69.
Trabalho realizado no Departamento de Genética e Biologia Molecular, Universidade Federal do Rio Grande do Sul e Serviço de Dermatologia, Universidade Federal de Ciências da Saúde de Porto Alegre / Santa Casa de Misericórdia de Porto Alegre, Porto Alegre, RS, Brasil.
