







Os pilomatricomas são tumores benignos originários da matriz capilar, os quais podem apresentar‐se como lesões solitárias ou, menos comumente, múltiplas. Quando múltiplos, podem estar associados a síndromes. A distrofia miotônica e a polipose adenomatosa familiar são as mais citadas na literatura. Há poucas publicações que relacionem esses tumores a outras síndromes genéticas. A síndrome de Rubinstein‐Taybi é um distúrbio autossômico dominante raro, caracterizado por deficiência intelectual e características dismórficas típicas. Há cinco casos publicados que relacionam pilomatricomas múltiplos à síndrome de Rubinstein‐Taybi, uma associação que ainda precisa ser esclarecida. Por esse motivo, relatamos os primeiros casos de pilomatricomas múltiplos em gêmeos monozigóticos com síndrome de Rubinstein‐Taybi típica.
Os pilomatricomas são tumores benignos pouco frequentes, originários da matriz capilar. Acometem principalmente a população pediátrica e localizam‐se com maior frequência na cabeça e no pescoço. Apresentam‐se clinicamente como nódulos de consistência firme ou pétrea, circunscritos, normocrômicos ou eritematosos, podem ser confundidos com cistos epidérmicos. Embora usualmente se apresentem como lesões solitárias, múltiplos pilomatricomas podem ocorrer em 2,4% a 5% dos casos.1–3
Pilomatricomas múltiplos podem ser esporádicos, familiares ou estarem associados a uma síndrome subjacente. A distrofia miotônica e as síndromes relacionadas à polipose adenomatosa familiar (PAF) são as mais citadas na literatura. Há relatos esporádicos desses tumores associados às síndromes de Turner, Kabuki, Sotos e Rubinstein‐Taybi2.
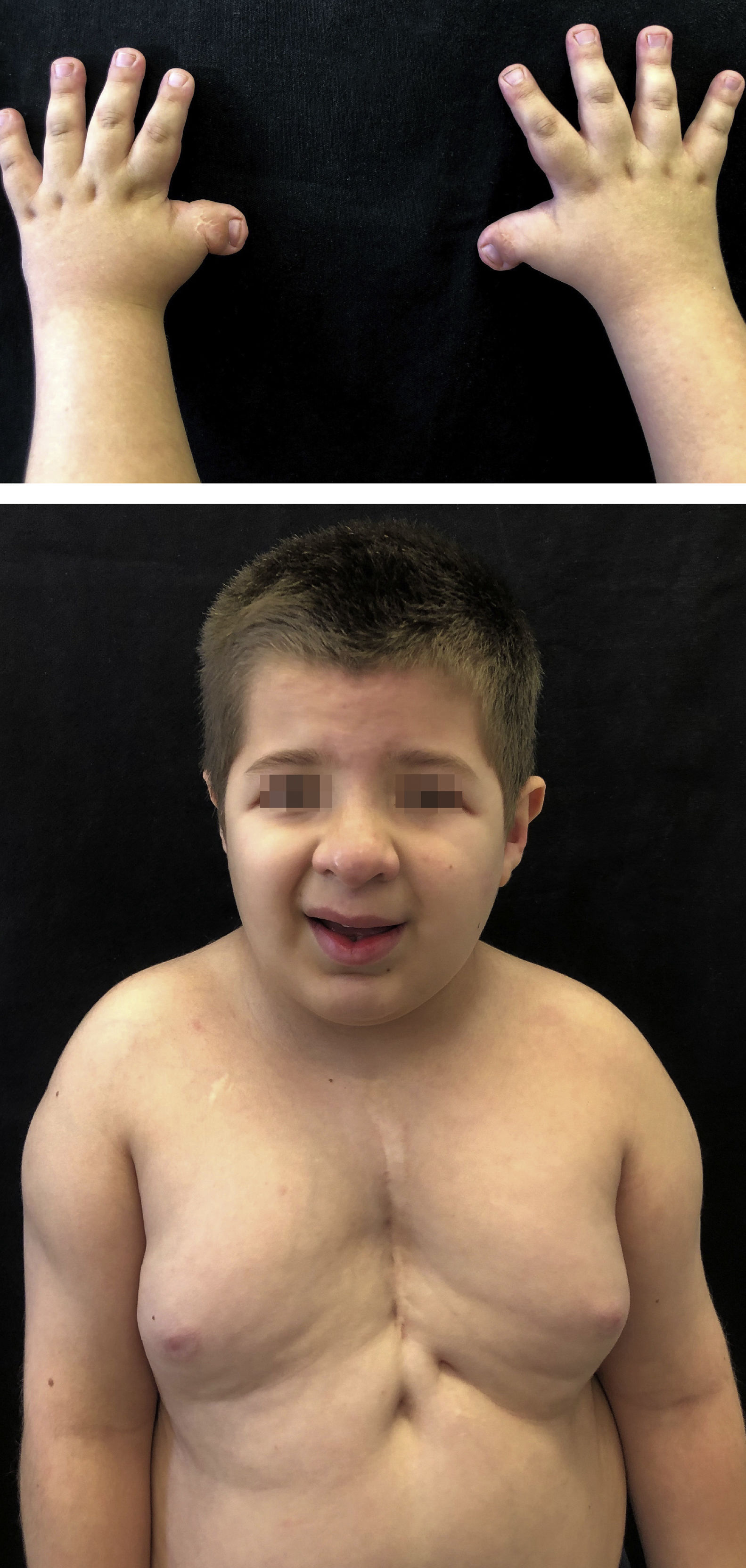
A síndrome de Rubinstein‐Taybi (SRT) é um distúrbio genético autossômico dominante raro caracterizado por retardo de crescimento pós‐natal, deficiência intelectual moderada a grave e uma ampla gama de características dismórficas típicas. Polegares e háluces largos e angulados são uma característica distintiva da síndrome. As anomalias faciais incluem fendas palpebrais oblíquas, pirâmide nasal alta e alongada e micrognatia. Malformações cardíacas, alterações dentárias e criptorquidia são comuns4. Os achados dermatológicos mais comuns incluem hemangiomas, hipertricose, braquioníquia e tendência à formação de queloides1.
Embora aproximadamente 60% dos casos estejam associados a mutações no gene CREBBP ou EP300, a etiologia da SRT é heterogênea e pouco definida1,5.
Há poucos casos que descrevem a associação de pilomatricomas à SRT e há dúvidas se essa se deve ao acaso6. De acordo com recente revisão da literatura, existem nove casos relatados até o momento, dos quais cinco são de lesões múltiplas2. Relatamos o primeiro caso de pilomatricomas múltiplos em gêmeos monozigóticos com SRT típica.
Relato dos casosGêmeos monozigóticos de 8 anos, com atraso no desenvolvimento neuropsicomotor, fendas palpebrais oblíquas, micrognatia discreta, palato ogival, hélice auricular proeminente, hipertelorismo mamilar e hipertricose leve sobre a coluna dorsal e ombros. Ambos tinham história prévia de criptorquidia e de polegares curtos e largos, com desvio radial corrigido cirurgicamente. O gêmeo 1 havia sido submetido à cirurgia cardíaca para correção de comunicação interventricular e tinha história prévia de hemangiomas occipital, frontal e supramamilar, os quais haviam regredido espontaneamente (fig. 1). O gêmeo 2 apresentava estenose de aqueduto cerebral e polidactilia, também previamente corrigida. O exame de cariótipo de ambos os pacientes era normal (46, XY) e, frente aos achados fenotípicos típicos, foram diagnosticados com SRT pela equipe da genética.
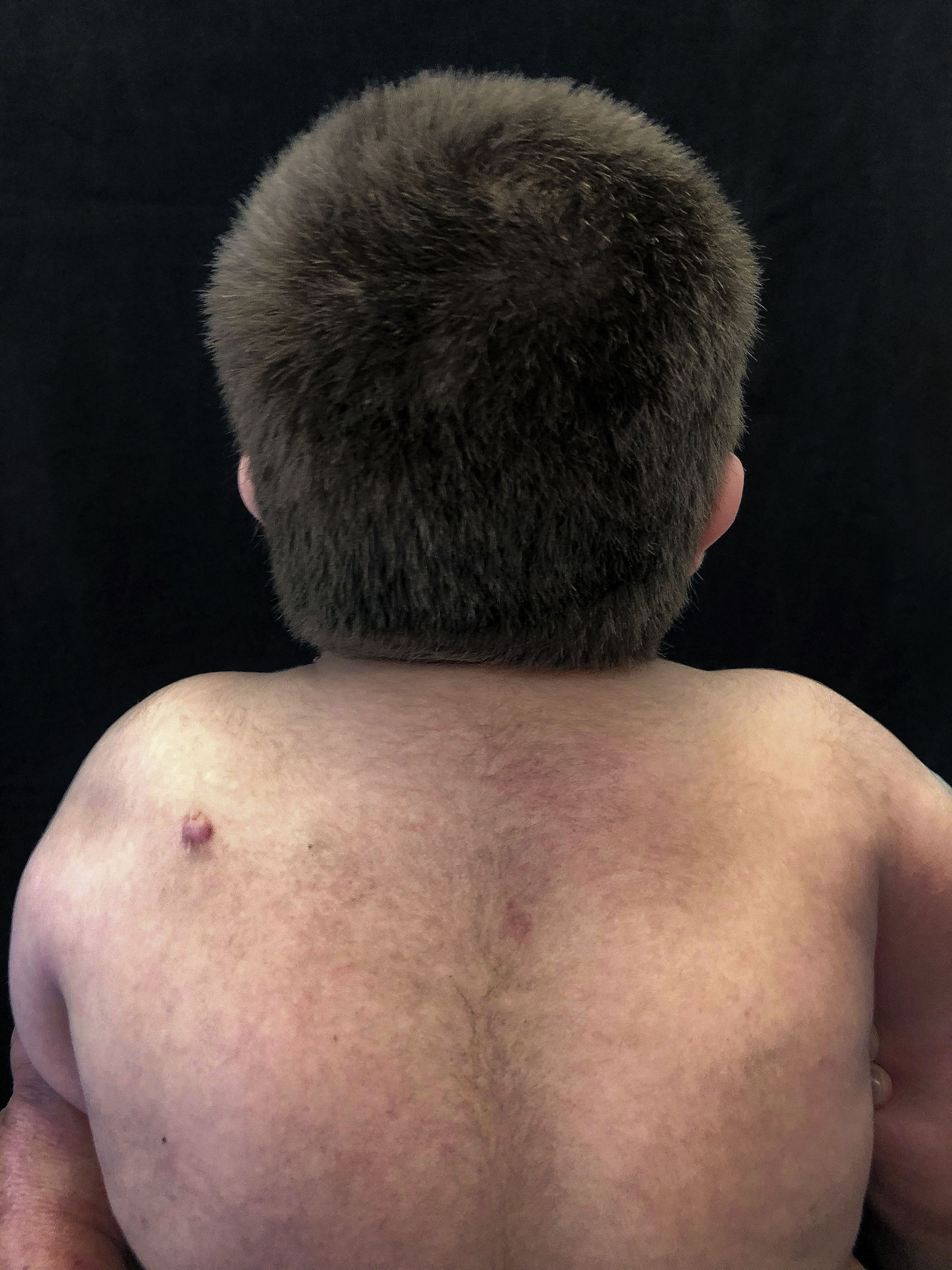
Em consulta dermatológica, o gêmeo 1 apresentava lesão nodular normocrômica, assintomática, na região escapular esquerda. Fez‐se ultrassonografia, que mostrou imagem nodular ecogênica, emitia sombra acústica posterior, com 1,4cm no maior diâmetro. Nas consultas subsequentes, em um período de dois anos, ambos os pacientes desenvolveram múltiplas lesões nodulares semelhantes no couro cabeludo (total de cinco lesões no gêmeo 1 e quatro no gêmeo 2). Duas delas foram excisadas, uma localizada no couro cabeludo e outra na região escapular (fig. 2), ambas evidenciavam histologicamente proliferação nodular composta por células basaloides matriciais e células fantasma (figs. 3 e 4), achados compatíveis com o diagnóstico de pilomatricomas. As demais lesões foram acompanhadas clinicamente. Os pacientes não tinham histórico familiar de pilomatricomas.
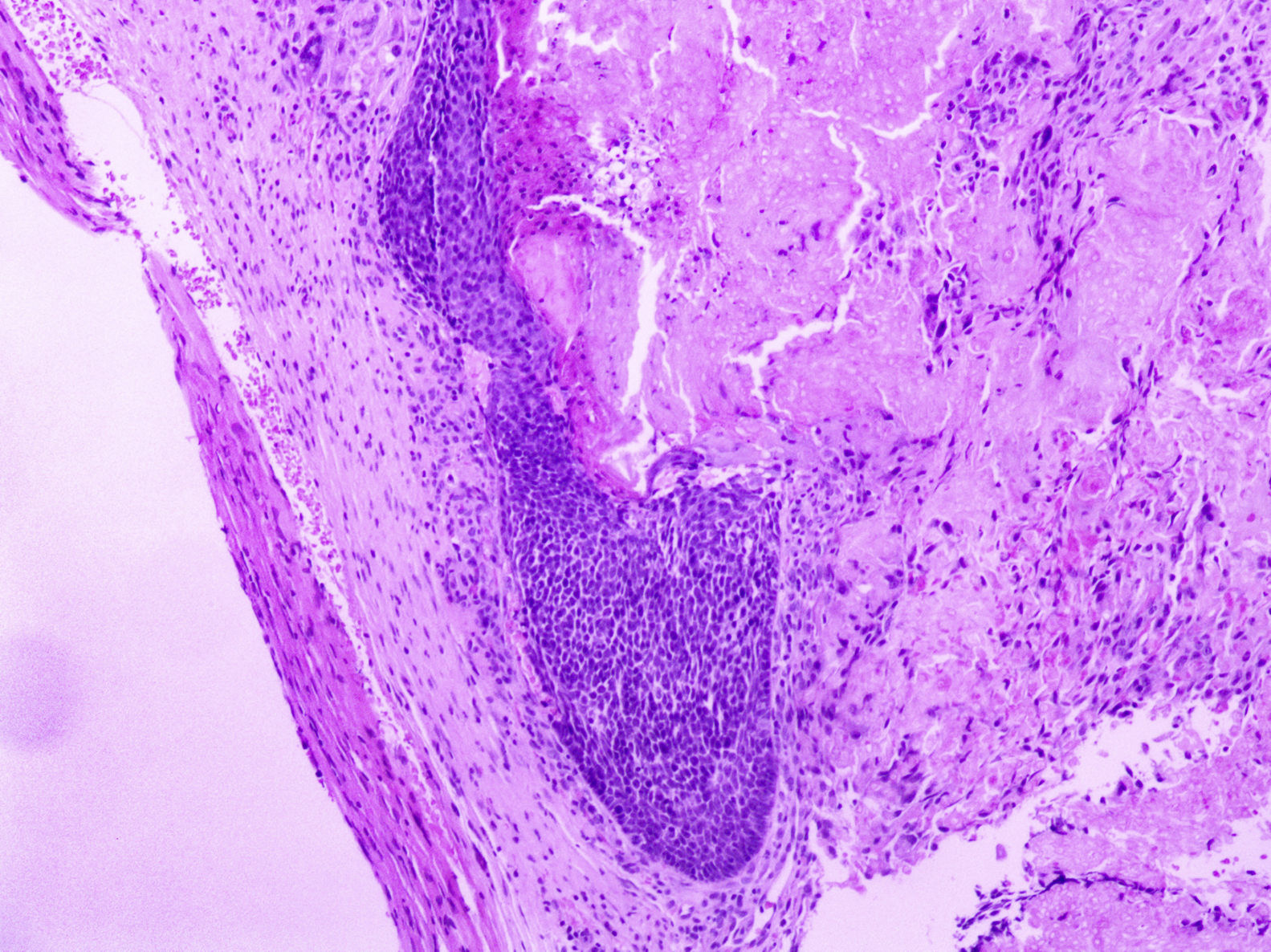
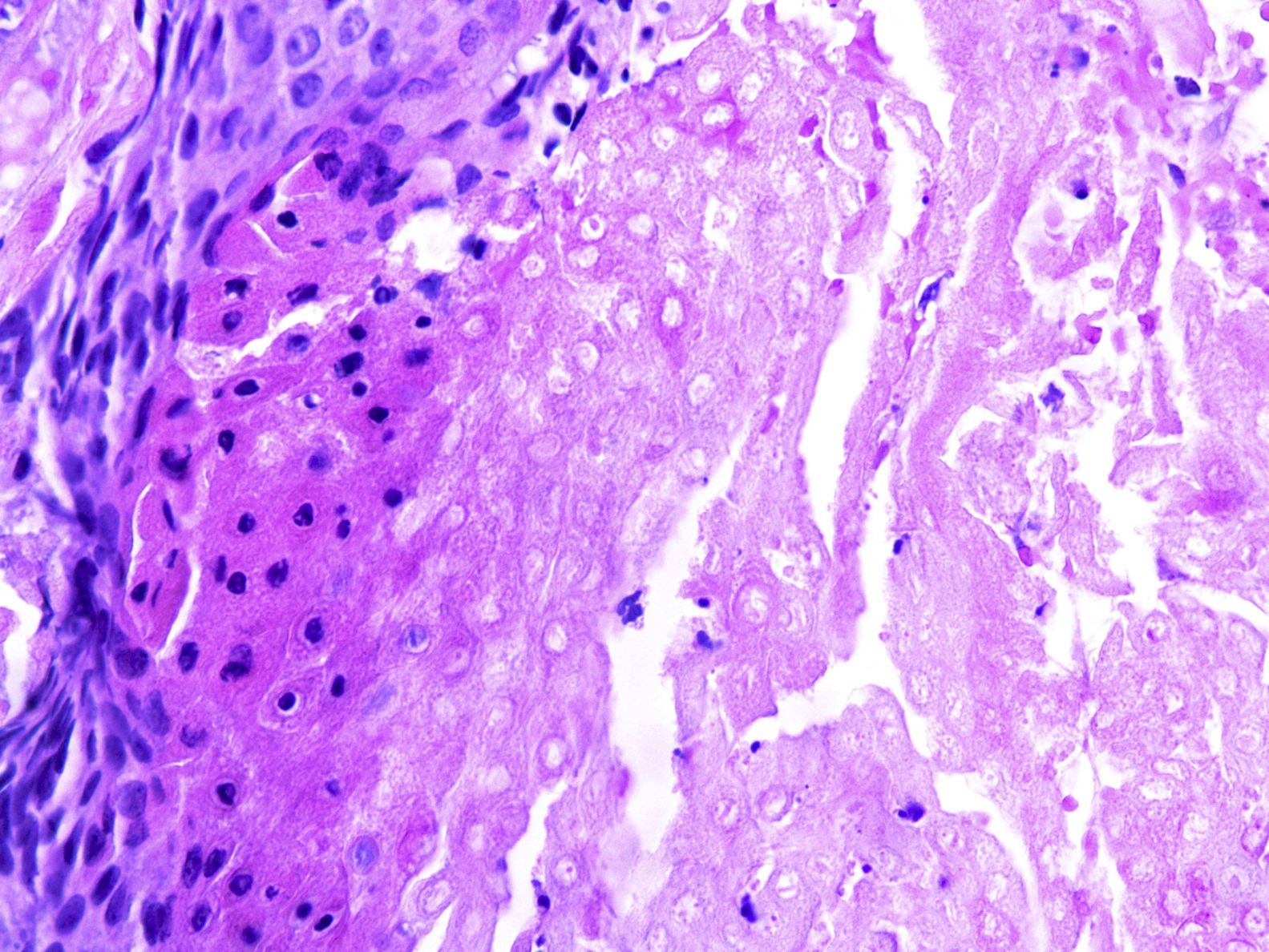
Os pilomatricomas geralmente são lesões solitárias benignas. No entanto, esses tumores podem ter apresentação múltipla e, embora possam ocorrer em indivíduos saudáveis, em pacientes com esse achado recomenda‐se coletar histórico familiar detalhado e descartar síndromes associadas7.
Distrofia miotônica e PAF são as síndromes mais conhecidamente relacionadas com múltiplos pilomatricomas. A associação entre SRT e pilomatricomas foi publicada pela primeira vez em 19946 e, desde então, mais oito casos foram descritos. No entanto, apenas cinco deles relacionavam múltiplos pilomatricomas com a síndrome.2
Uma revisão de 2019 sobre síndromes associadas a múltiplos pilomatricomas evidenciou que os casos de pacientes não sindrômicos tendem a apresentar menor número de pilomatricomas quando comparados aos sindrômicos2. Enquanto 4,5% dos indivíduos não sindrômicos desenvolveram mais de cinco pilomatricomas, 46,3% dos pacientes sindrômicos desenvolveram seis ou mais tumores. No entanto, embora a relação entre múltiplos pilomatricomas e síndrome subjacente se fortaleça à medida que o número de pilomatricomas aumenta, algumas síndromes podem apresentar apenas uma ou duas lesões2. A revisão dos casos de SRT descritos até 2019 mostrou que o número de pilomatricomas associados a essa síndrome é bastante variado. De nove casos, quatro tiveram dois a cinco pilomatricomas, um teve mais de 10 e quatro tiveram pilomatricomas solitários1,2,5,6,8–10. Além disso, uma série de quatro casos publicados em 1998 mostrou que, nesse grupo de pacientes, a idade média de surgimento dos tumores não foi mais precoce do que em pacientes saudáveis, nos quais a maioria das lesões surge entre 8 meses e 10 anos9.
A etiologia do pilomatricoma na SRT ainda precisa ser elucidada. Mutações em dois genes, CREBBP e EP300, foram identificadas em indivíduos afetados. Ambos codificam proteínas homólogas que atuam como coativadores da transcrição e, durante a organogênese, CREBBP é expresso em tipos celulares específicos do coração, vasculatura, pele, pulmões e fígado em desenvolvimento. Em 2016, foi relatado o primeiro caso de SRT com múltiplos pilomatricomas diagnosticado por análise de mutação CREBBP. No entanto, a correlação entre o genótipo CREBBP e o desenvolvimento de múltiplos pilomatricomas ainda precisa ser esclarecida, uma vez que também existem relatos de casos que descrevem a mutação CREBBP em pacientes com SRT sem pilomatricomas ou com lesão solitária5.
Os dados sobre pilomatricomas na SRT ainda estão limitados a alguns relatos de casos. Na revisão da literatura feita, este é o primeiro relato de pilomatricomas múltiplos em gêmeos com SRT, o que reforça a associação entre essas duas entidades. Os mecanismos moleculares que levam os pacientes com SRT a uma maior suscetibilidade a pilomatricomas necessitam ser estudados.
Em alguns casos, o surgimento de pilomatricomas pode oferecer uma oportunidade para o diagnóstico de SRT. A abordagem terapêutica de múltiplas lesões no RTS ainda não está estabelecida. Nos pacientes apresentados, optou‐se pela abordagem conservadora, com acompanhamento dos pacientes sem intervenção.
Suporte financeiroNenhum.
Contribuição dos autoresAna Laura Andrade Bueno: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Maria Emilia Vieira de Souza: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Carla Graziadio: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Ana Elisa Kiszewski: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito;, obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Conflitos de interesseAna Laura Andrade Bueno, Maria Emilia Vieira de Souza e Carla Graziadio declaram não haver conflitos de interesse. Ana Elisa Kiszewski declara conflito de interesses com a empresa Johnson & Johnson.
Como citar este artigo: Bueno ALA, Souza MEV, Graziadio C, Kiszewski AE. Multiple pilomatricomas in twins with Rubinstein‐Taybi syndrome. An Bras Dermatol. 2020;95:619–22.
Trabalho realizado no Serviço de Dermatologia, Universidade Federal de Ciências da Saúde de Porto Alegre e na Unidade de Dermatologia Pediátrica, Hospital da Criança Santo Antonio, Irmandade da Santa Casa de Misericórdia de Porto Alegre, Porto Alegre, RS, Brasil.
