






Hypereosinophilic syndrome is defined as persistent eosinophilia (>1500/μL for more than six months) associated with organ involvement, excluding secondary causes. It is a rare, potentially lethal disease that should be considered in cutaneous conditions associated with hypereosinophilia. We report a case of erythroderma as a manifestation of hypereosinophilic syndrome. A 36-year-old male with no comorbidities presented progressive erythroderma, pruritus, peripheral neuropathy, and eosinophilia in the previous seven months. No mutations were found in FIP1L1/PDGFRA. Patient experienced rapid remission in response to oral prednisone and hydroxyurea. Cutaneous manifestations may be the only evidence of hypereosinophilic syndrome. Genotyping excludes myeloproliferative disease, thereby orienting treatment and prognosis.
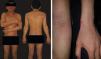
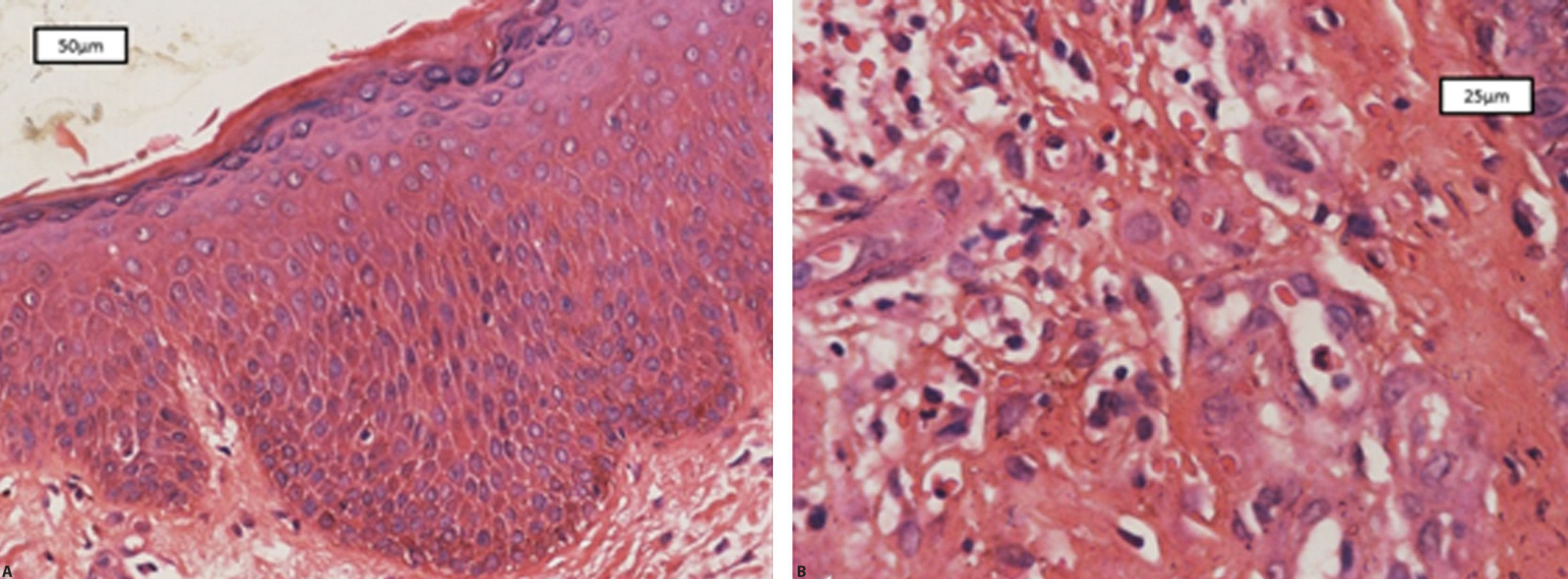
Patient was a 36-year-old Brazilian male physician of Japanese descent with no comorbidities or atypical signs. Six years previously he presented intermittent erythematous papules on the trunk and face, in addition to pruritus aggravated by heat or alcohol intake. Six months previously he evolved with erythroderma, treated with cyclosporine (3mg/kg/d), hydration, and topical corticosteroids, with partial improvement. On physical examination: erythroderma, facial infiltration, and infiltrated erythematous plaques (mainly cubital and popliteal), xeroderma, thickening of the greater auricular, fibular, radial, and ulnar nerves with reduced strength in hands (Figure 1). Anatomical pathology of the skin revealed acanthosis, spongiosis, and superficial lymphocytic infiltrate with eosinophils (Figure 2). Complete blood count: eosinophilia 2,812U/μL; IgE 1,164 kU/L (ULN 114kU/L), and DHL 596U/L (ULN 225U/L). Ultrasound of the wrists showed thickening of the median nerve with normal morphology of the carpal tunnel. Normal echocardiogram, and abdominal tomography showed periaortic lymph nodes slightly increased in number.
Hypereosinophilic syndrome (HES) is characterized by persistently elevated peripheral eosinophil count (> 1,500/μL) associated with symptomatic organ involvement, excluding secondary causes of eosinophilia (Chart 1).1 It is classified as myeloproliferative (HES-M), secondary or reactive (HES-L), and idiopathic (HES-I) (Chart 2)2
Diagnostic criteria for hypereosinophilic syndrome (HES)
| Original diagnostic criteria, Chusidet al.1 | Modification to diagnostic criteria for HES by Simonet al.1 | Comments |
|---|---|---|
| 1. Peripheral eosinophilia > 1500/μL persisting for more than six consecutive months. | Persistent eosinophilia. Arbitrary value of 1500/μL is not mandatory | Presence of tissue infiltration with eosinophils leading to tissue damage is more important than setting a laboratory value |
| 2. Rule out secondary causes of hypereosinophilia after extensive clinical workup | Peripheral eosinophilia should be confirmed, but its persistence for more than six months is no longer mandatory | In symptomatic patients with confirmed eosinophilia and organ involvement, proceed to treatment to prevent organ damage; any duration is significant, even less than six months |
| 3. Symptomatic organ involvement secondary to hypereosinophilia | Signs and/or symptoms of organ involvement are not mandatory | At the time of diagnosis, it is very difficult to predict evolution in asymptomatic patients. Some may be asymptomatic at first evaluation and either remain asymptomatic or evolve due to tissue damage or organ involvement |
Main features of hypereosinophilic syndrome (HES)
| Subtypes | Characteristics | Treatment | Prognosis |
|---|---|---|---|
| Myeloproliferative (HES-M) | Tyrosine kinase activation by FIP1L1 and PDGFRA gene fusion, eosinophilia due to clonal proliferation of myeloid precursors | Little response to systemic corticosteroids. More response to hydroxyurea, interferon, and chemotherapy – busulfan, chlorambucil, and vincristine. Imatinib is the drug of choice for patients with positive F/P mutation | Survival depends on severity of tissue injury, especially the degree of cardiac involvement. Risk of development of malign myeloproliferative disease |
| Idiopathic (HES-I) | T-cell polyclonal expansion unregulated production of Th2 cytokines (IL-5 and/or IL-3) | Good response to systemic corticosteroids alone or in combination with hydroxyurea, mepolizumab, interferon-α, or tofacitinib | Chronic, benign condition with indolent clinical course. Occasional development of lymphoma |
| Lymphocytic/Secondary (HES-L) | Polyclonal expansion of anomalous T-cells (CD3-CD4+) and unregulated production of Th2 cytokines (especially IL-5) | Adequate control with systemic corticosteroids alone or in combination with hydroxyurea, mepolizumab, or tocilizumab | Chronic recurrent. Occasional development of lymphoma |
HES is a rare disease (5,000 cases/year in the United States of America), potentially fatal, and the organs most frequently involved are skin, lungs, intestine, heart, kidneys, eyes, and peripheral nervous system. The most serious complication is cardiac, which should be investigated in all cases. In adults, the syndrome is more common in males (ratio 9:1), and mean age at onset is 50 years. Investigation of organ involvement and sub-classification are decisive for treatment and prognosis.3,4
Cutaneous manifestations of HES require differential diagnosis with urticaria, pruritus sine materia, mycosis fungoides, cutaneous adverse drug reactions, contact dermatitis, and atopic dermatitis. Dermatologists should be alert to pruritic erythematous papules, urticaria, angioedema, dermographism, oral and genital ulcers, centrifugal annular erythema, acral bullae, and erythroderma. Histopathologic examination of the skin lesion is usually nonspecific, with viable eosinophilic infiltration.4-7
In an endemic country like Brazil, a presentation with polyneuropathy requires ruling out leprosy, which is unlikely in this case because of the pruritus and histology.
In this case, treatment began with prednisone 1mg/kg/day, with important improvement in the skin lesions, eosinophilia (580/μL), and pruritus after 30 days. Bone marrow biopsy did not show neoplastic infiltration, FISH cytogenetic test did not reveal deletion or translocation of the FIP1L1 and PDGFRA genes, JAK2 mutation was negative, and immunophenotyping did not show clonal lymphocyte populations. Adjuvant therapy with hydroxyurea was scheduled in conjunction with the Department of Hematology.
The case was classified as HES-I based on the absence of lymphocytic phenotypical abnormalities or genetic alterations. Presence of peripheral neuropathy, absence of atopic signs, and worsening of the clinical picture with alcohol intake were characteristic. High IgE levels, good response to corticosteroid therapy, and low serum tryptase level and eosinophil count < 100,000/μL are associated with better prognosis. Therapeutic success in HES-I has been reported with tofacitinib and mepolizumab.4,8-10
Diagnosis of HES is often delayed due to its pleomorphic dermatological manifestations and insidious evolution. It is thus crucial that in all cases of erythroderma, HES should be considered as a differential diagnosis.6,7
Financial support: None.
Conflict of interest: None.
