





A síndrome de Rothmund‐Thomson (SRT) é distúrbio autossômico recessivo raro, caracterizado por lesão facial (poiquilodermia é característica diagnóstica importante), retardo de crescimento, cabelo/cílios/sobrancelhas esparsos, cataratas juvenis, anormalidades esqueléticas, defeitos ósseos do antebraço ou mão e uma predisposição ao câncer. Existem duas formas clínicas: o tipo I, caracterizado por poiquilodermia, displasia ectodérmica e catarata juvenil de etiologia desconhecida, e o tipo II, caracterizado por poiquilodermia, defeitos ósseos congênitos, maior frequência de neoplasias malignas (principalmente osteossarcoma) e mutação no gene RECQL4 (8q24.3).1 Até o momento, foram relatados cerca de 400 casos.
Os autores relatam caso de poiquilodermia e retardo de crescimento em uma menina chinesa com duas mutações RECQL4 em um novo arranjo heterozigótico composto (c.2492_2493del e c.1391‐2A>C) registrado por triagem mutacional, o primeiro relato em SRT.
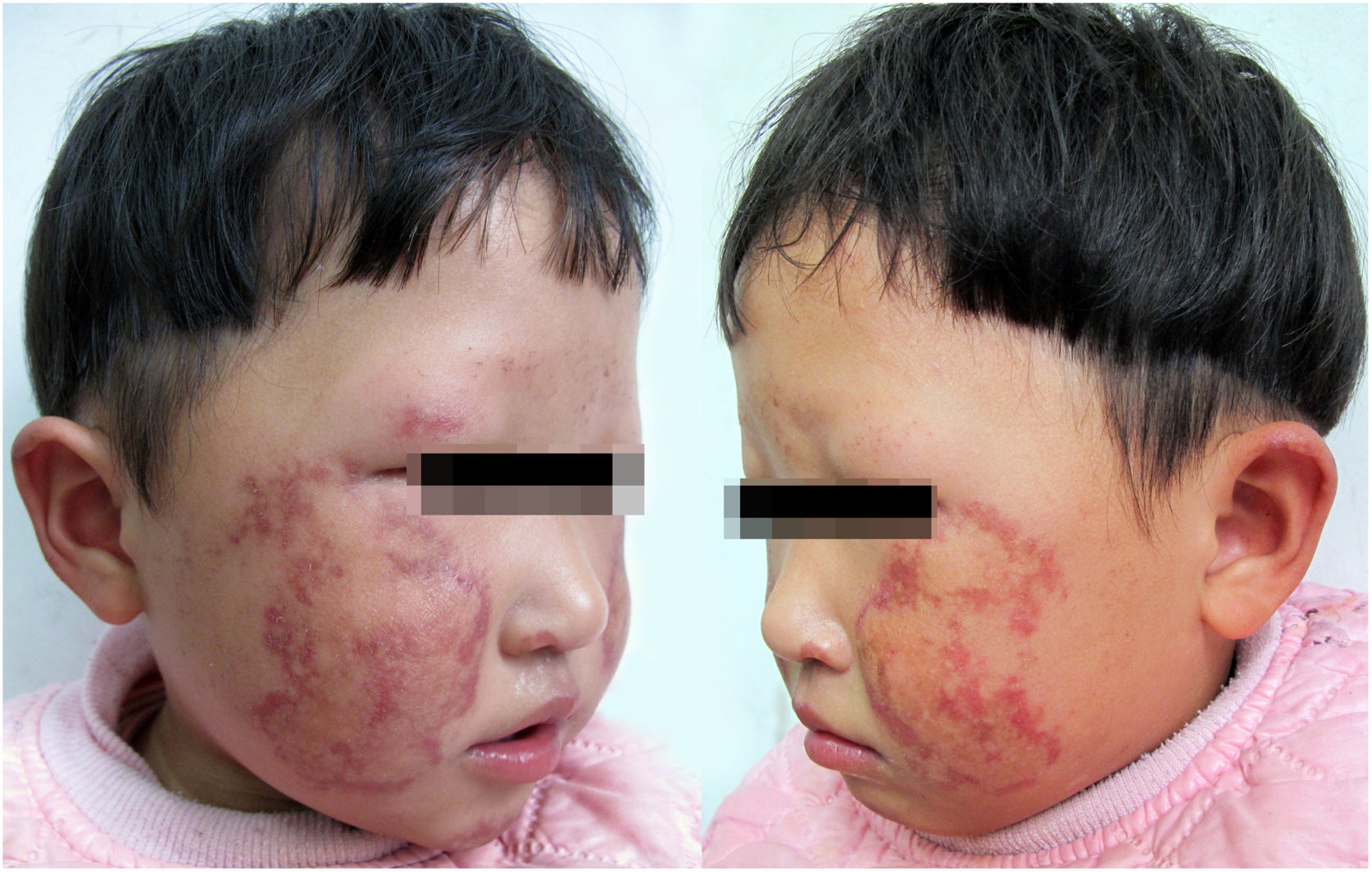
A paciente é menina de 2 anos com poiquilodermia bilateral na face e orelhas. Os pais relataram que a criança apresentava eritema, edema e formação de bolhas na face bilateralmente desde os 6 meses, que gradualmente evoluíram para hipo‐ e hiperpigmentação reticulada. A paciente também apresentou afinamento das sobrancelhas, fotossensibilidade e problemas gastrintestinais, inclusive vômitos ou diarreia crônica. Nenhum dos pais ou a irmã de 5 anos apresentou os mesmos sintomas. A paciente nasceu a termo, com leve anormalidade nos dedos dos pés. No entanto, ganho de peso lento, baixa estatura e retardo dentário foram observados no exame físico. O exame dermatológico encontrou despigmentação, hiperpigmentação, atrofia puntiforme e telangiectasia bilateralmente na face e nas orelhas (fig. 1). Quando a paciente tinha um ano, sua densidade mineral óssea foi avaliada e considerada baixa. Capacidade cognitiva, testes oftalmológicos e outros resultados de exames estavam dentro dos limites normais e nenhuma outra alteração foi encontrada.
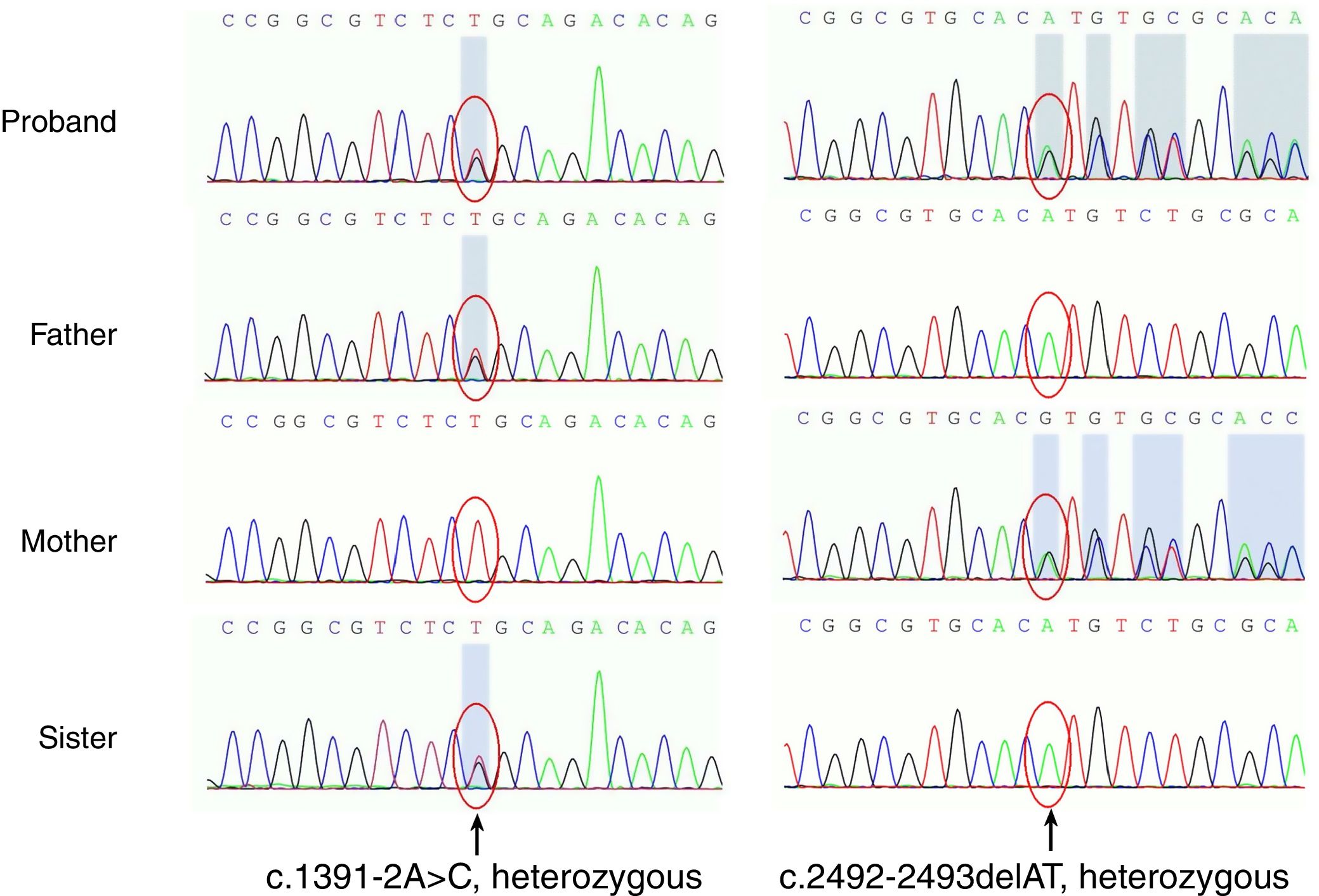
Para o diagnóstico molecular diferencial de poiquiloderma, foi feito o sequenciamento de exoma direcionado. A triagem mutacional para BLM, o gene defeituoso na síndrome de Bloom e em outras doenças relacionadas à poiquilodermia, foi negativa. O sequenciamento genético revelou duas mutações heterozigóticas no gene RECQL4 (fig. 2). Uma delas é uma mutação localizada no éxon 9, que consiste na alteração de uma adenina por uma citosina (c.1391‐2A>C), que também foi observada no pai e na irmã, ambos não afetados. Essa mutação não foi relatada, mas deve-se assumir um possível efeito sobre a proteína por meio de uma variante aceitadora de splicing. No outro alelo, a mutação foi uma deleção de dois nucleotídeos encontrados no éxon 16 (c.2492_2493delAT) que produz um deslocamento de quadro (p.His831Argfs); essa mutação é rara e foi mais recentemente avaliada por Kitao et al.2 Essa mutação foi observada na mãe, não afetada. Essas duas mutações, respectivamente, são de origem materna e paterna e foram denominadas mutações heterozigóticas compostas de acordo com a lei de herança autossômica recessiva. Sua irmã apresenta apenas c.1391‐2A>C, uma mutação heterozigótica, e teoricamente não apresentará sintoma.
A paciente tem sinais clínicos como poiquilodermia, sobrancelhas esparsas, baixa estatura, anormalidade dentária e anormalidade esquelética leve, como descrito na literatura.3,4 Até o momento, a paciente não apresenta catarata ou neoplasia. Ao contrário de outros casos relatados na literatura, a lesão não afeta suas extremidades e os autores consideram que a paciente é jovem demais para mostrar todos os sintomas. A SRT foi diagnosticada de acordo com a lesão e a mutação típica do gene RECQL4; aconselhou‐se evitar a exposição solar e fazer exames anuais para olhos, pele e ossos.
As novas mutações heterozigóticas compostas do RECQL4 apresentadas por essa paciente são as primeiras relatadas na SRT. A perda da função da proteína RECQL4 foi observada em aproximadamente dois terços dos pacientes com SRT e está associada ao risco de osteossarcoma.5 São necessários mais estudos funcionais para confirmar o efeito prejudicial às proteínas. A poiquilodermia é manifestação de muitas doenças sistêmicas, como lúpus eritematoso, síndrome de Bloom, síndrome de Kindler e disqueratose congênita. O resultado do teste genético é importante para um diagnóstico definitivo e orientação quanto à futura procriação para a família dos pacientes.
Suporte financeiroNenhum.
Contribuição dos autoresXinyue Zhang: Elaboração e redação do manuscrito.
Songmei Geng: Aprovação da versão final do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados.
Yi Zheng: Revisão crítica da literatura.
Conflitos de interesseNenhum.
Como citar este artigo: Zhang X, Geng S, Zheng Y. Rare presentation of Rothmund‐Thomson syndrome with novel compound heterozygous mutations of the RECQL4 gene. An Bras Dermatol. 2020;95:538–40.
Trabalho realizado no Departamento de Dermatologia, Second Affiliated Hospital, Xi’An Jiaotong University, Shaanxi, China.
