






As epidermólises bolhosas (EB) constituem desordens bolhosas hereditárias nas quais as bolhas surgem espontaneamente ou são desencadeadas por mínimos traumas; essa denominação foi sugerida por Koebner em 1886. As EB são divididas em quatro grandes grupos (simples, juncionais, distróficas e a síndrome de Kindler) e em diversos fenótipos clínicos distintos, de acordo com o nível de clivagem e as características clínicas e moleculares.1,2
A epidermólise bolhosa simples com pigmentação moteada (EBS‐PM) é um raro subtipo da epidermólise bolhosa simples (EBS) (Online Mendelian Inheritance in Man [OMIM] no. 131960). Caracteriza‐se por bolhas não cicatriciais principalmente nas extremidades distais e por hiperpigmentação moteada progressiva. Até 2013, apenas 15 famílias e oito casos esporádicos haviam sido relatados, segundo dados do Hospital Infantil Universitário Niño Jesus, de Madrid, o que nos motivou a fazer este relato.2
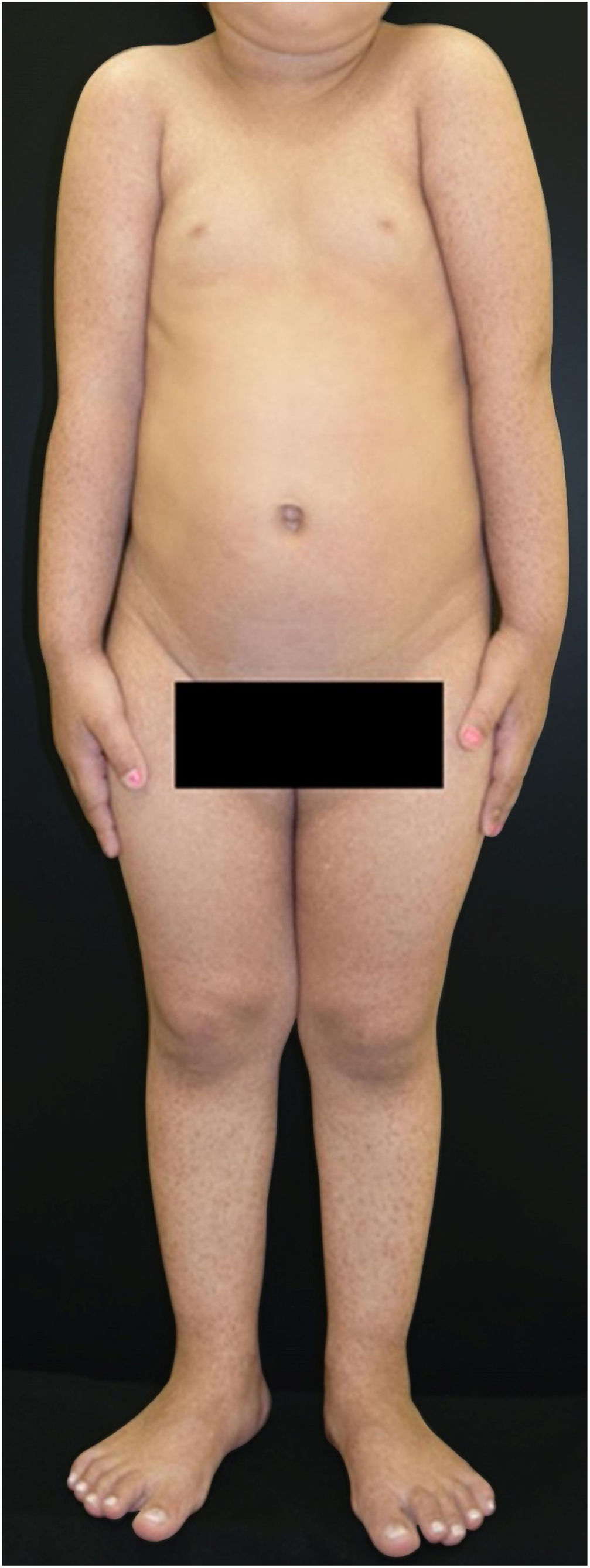
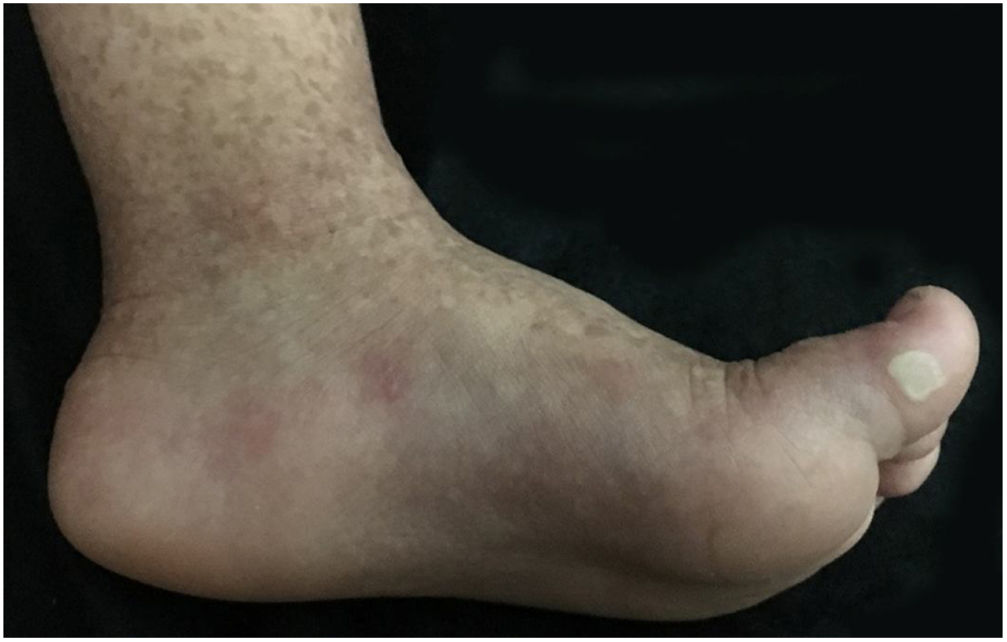
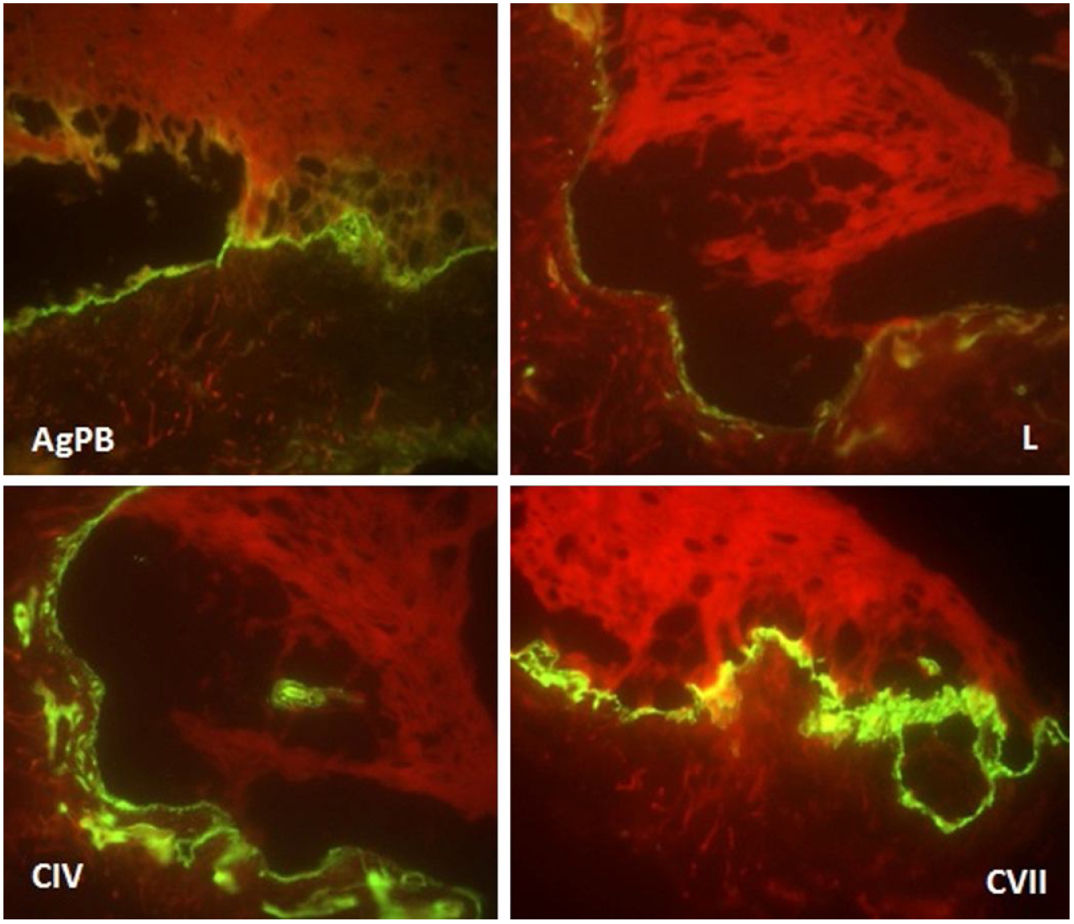
Paciente do sexo feminino, 2 anos, fototipo III, com história de bolhas na pele desde o nascimento. Ao exame dermatológico apresentava bolhas dessecadas nos pés e máculas hiper e hipocrômicas dispersas pelo tegumento com aspecto moteado (figs. 1 e 2). Pápulas normocrômicas no dorso dos quirodáctilos e onicodistrofia também foram observadas. As bolhas apareciam de maneira espontânea ou após mínimos traumas, segundo o relato da mãe, e localizavam‐se principalmente nas extremidades distais dos membros. Aos 2 meses, iniciaram‐se as máculas hiper e hipocrômicas. A mãe também referia episódios de mucosite oral leve. Foi feita biópsia de bolha recente e o material foi levado para imunomapeamento (fig. 3), o qual resultou em EBS. Considerando‐se a clínica da paciente e o achado laboratorial, concluiu‐se pelo diagnóstico de EBS‐PM, possível caso esporádico, frente à ausência de história familiar para EB ou outra dermatose bolhosa até o momento. A paciente está em acompanhamento clínico ambulatorial. Orientações familiares foram feitas a fim de reduzir o surgimento de novas bolhas e melhorar a convivência da paciente com sua genodermatose.
Descrita pela primeira vez em 1979 por Fischer e Gedd‐Dahl, a EBS‐PM tem início na infância e origem genética. É uma EBS basal causada por uma mutação no gene KRT5 que codifica a citoqueratina 5. Ocorre mais comumente devido a uma mutação p24L heterozigótica circunstancial no domínio V1 não helicoidal da citoqueratina 5.1,2
O diagnóstico dessa dermatose é baseado em achados clínicos característicos, história familiar, imunomapeamento e/ou microscopia eletrônica de transmissão, bem como análise molecular ou da mutação, quando possível.2
Clinicamente, caracteriza‐se por bolhas não cicatriciais principalmente nas extremidades distais e por hiperpigmentação moteada progressiva, a qual não ocorre no local das bolhas e frequentemente desaparece na vida adulta. Alguns casos podem ser acompanhados de máculas hipocrômicas, conforme observado em nossa paciente. Também há registro de hiperqueratose focal palmar e plantar. Pequenas pápulas verrucosas acrais, onicodistrofia e acometimento leve da mucosa podem ser observados durante a infância. Achados incomuns incluem fotossensibilidade e desordens dentárias (cáries).2
O diagnóstico diferencial de EBS‐PM inclui outras formas de EBS (principalmente tipo herpetiforme de Dowling‐Meara), síndrome Kindler, displasia ectodérmica de Naegeli‐Franceschetti‐Jadassohn (NFJ), outras formas de discromia, doença de Dowling‐Degos e até mesmo casos atípicos da doença de Darier com mutações em ATP2A2.2–4
Frente à hipótese clínica de EB e para determinar o plano de clivagem, o imunomapeamento ou a microscopia eletrônica de transmissão deve ser feito. O imunomapeamento tem precisão diagnóstica semelhante à da microscopia eletrônica de transmissão, com a vantagem de execução e leitura mais simples e rápida. Está associado ao uso de anticorpos monoclonais e pode ser considerado uma técnica de imunofluorescência indireta. Na EBS, a clivagem ocorre na camada basal (intraepidérmica). O depósito da fluorescência no assoalho da bolha (lado dérmico) é observado com todos os marcadores antigênicos (antígeno do penfigoide bolhoso, laminina, colágeno IV e VII), como observado neste caso.5
A análise ultraestrutural das áreas pigmentadas nessa forma de EBS demonstra abundantes melanossomas maduros dentro das células basais.2
Assim, relatamos um caso raro de EBS‐PM, possivelmente esporádico. Ressaltamos a raridade desse subtipo de EBS e suas características clínicas marcantes que favorecem diagnósticos futuros e destacamos seu caráter benigno, com lesões não cicatriciais ou deformantes e com regressão da hiperpigmentação na vida adulta.
Suporte financeiroNenhum.
Contribuição dos autoresFlávia Regina Ferreira: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica da literatura; revisão crítica do manuscrito.
Carolina Fernandes Pereira: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; revisão crítica da literatura.
Juliana Carvalho Moretto: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; revisão crítica da literatura.
Mariana Patriota Naville: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito; revisão crítica da literatura.
Conflitos de interesseNenhum.
Como citar este artigo: Ferreira FR, Pereira CF, Moretto JC, Naville MP. Sporadic form of epidermolysis bullosa simplex with mottled pigmentation. An Bras Dermatol. 2020;95:536–8.
Trabalho realizado no Hospital Universitário de Taubaté, Taubaté, SP, Brasil.
